4+3 cycloaddition
A [4+3] Cycloaddition is a cycloaddition between a 4 atom pi-system and a 3 atom pi-system to form a seven-membered ring. Allyl or oxyallyl cations (propenylium-2-olate) are commonly used 3 atom pi-systems, while a diene (such as butadiene) plays the role of the 4 atom pi-system. It represents one of the relatively few synthetic methods available to form seven-membered rings stereoselectively in high yield.[1] The reaction is a topic academic research.
Introduction
Symmetry-allowed [4+3] cycloaddition is an attractive method for the formation of historically difficult-to-access seven-membered rings. Neutral dienes and cationic allyl systems (most commonly oxyallyl cations) may react in a concerted or stepwise fashion to give seven-membered rings. A number of dienes have been employed in the reaction, although cyclic, electron-rich dienes such as those found in the pyrrole and furan ring systems are the best 4π systems for this process. Intramolecular variants are also efficient.[2]
(1)

Recent developments have focused on expanding the scope of enantioselective [4+3] cycloadditions and the range of conditions available for generating the key oxyallyl cation (propenylium-2-olate) intermediate.
Mechanism and Stereochemistry
Prevailing Mechanism
Oxyallyl cations (propenyliumolates) may be generated under reductive, mildly basic, or photolytic conditions. Reduction of α,α'-dihalo ketones is a very popular method for generating symmetric oxyallyl cations.[3] After formation of a metal enolate, dissociation of halide generates a positively charged oxyallyl intermediate. This electron-deficient 2π component reacts with electron-rich dienes to give cycloheptenones. Cyclic dienes fare better than the corresponding acyclic dienes because in order to react, the diene must be in the s-cis conformation in the presence of the short-lived oxyallyl cation—cyclic dienes are locked in this reactive conformation.
(2)

Substituents at the 1 and 3 positions are usually required to stabilize the oxyallyl cation and prevent isomerization to cyclopropanones and allene oxides.[3] In most cases, an excess of the diene is employed to prevent isomerization of the oxyallyl cation intermediate. Increasing the covalent character of the metal-oxygen bond (by, for instance, employing iron carbonyl reducing agents instead of sodium) also stabilizes the oxyallyl cation, leading to cleaner reactions. Strongly electrophilic allyl cations tend to give products of electrophilic substitution rather than cycloaddition.[4]
The cycloaddition itself may be either concerted or stepwise, depending on the nature of the oxyallyl intermediate and the reaction conditions.[5] Concerted reactions taking place under reductive conditions usually exhibit low regioselectivity due to somewhat indiscriminate frontier orbital control; however, stepwise (or at least asynchronous) reactions under basic conditions do exhibit moderate regioselectivity (attributed to initial formation of a bond between the less sterically hindered ends of the pi systems).
Stereochemistry
Stereochemical control in the [4+3] cycloaddition is not as strict as in the Diels-Alder reaction, because the former often proceed through stepwise, polar pathways. Even when the reaction is concerted, complications may arise due to conformational dynamics in the oxyallyl component, which can exist in "W," "U," or "sickle" forms. Generally, however, the "W" form dominates. Even so, two stereochemically distinct transition states are possible: a chair-like, "extended" TS which leads to a cis relationship between the bridging atom and oxyallyl substituents, and a boat-like, "compact" TS which leads to a trans relationship.
(3)
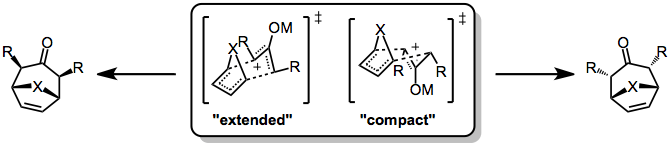
Which transition state is favored depends on both the 4π and 2π reacting partners. Reactions of cyclic dienes tend to favor the compact over the extended TS (this is particularly true for furan). In addition, the electrophilicity of the oxyallyl cation is related to the favorability of the extended transition state—more electrophilic cations (which possess more covalent metal-oxygen bonds) tend to favor the extended transition state, while less electrophilic cations favor the compact transition state.[6]
Scope and Limitations
Reduction of α,α'-dihalo ketones is an effective method for the generation of oxyallyl cations for cycloaddition. Reducing agents used include copper-bronze.,[7] iron carbonyl complexes.,[6] and copper/zinc[8] As mentioned previously, products exhibiting trans stereochemistry between the bridging atom and the oxyallyl substituents (resulting from the compact transition state) are generally favored.
(4)

α-Halo ketones with hydrogens at the α' position can also be transformed into oxyallyl cations under basic conditions. This usually requires highly polar media, and the use of a halophilic Lewis acid (such as Ag+) is sometimes necessary.[9]
(5)

Photochemical routes to oxyallyl cations generally result in the formation of a new covalent bond before the cycloaddition itself takes place. These reactions thus may lead to the formation of three new carbon-carbon bonds in a single operation.[10]
(6)

Intramolecular [4+3] cycloadditions are also possible, and often lead to interesting bridged architectures that are difficult to access by other methods. The product below, for instance, features a rare trans-bridging ketone.[11]
(7)

Synthetic Applications
A synthesis of Prelog-Djerassi lactone illustrates how stereocenters set during a [4+3] cyclization may be used later for stereochemical control.[12] The oxabicyclo[3.2.1]octane products of cycloadditions involving furan may be opened using a variety of methods[13]
(8)

Comparison with Other Methods
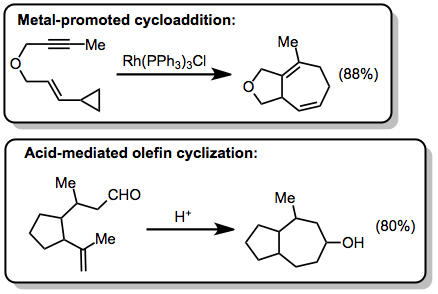
Compared to annulations that form five- and six-membered rings, annulations that form seven-membered rings are relatively rare. "Classical" methods that clip linear substrates together through the formation of a single carbon-carbon bond form seven-membered rings efficiently in some cases (cf. the acid-mediated olefin cyclization below[14]). Transition-metal catalyzed cycloadditions of vinylcyclopropanes are also useful for the formation of seven-membered rings[15]
(9)
Experimental Conditions and Procedure
Typical Conditions
Cycloadditions carried out under reductive conditions can generally be effected with commercially available reducing agents, although a few reducing agents require special preparation. Reductive reactions employing iron carbonyl complexes should be carried out in a well-ventilated fume hood, as free carbon monoxide may be released. The optimal conditions for base-mediated cycloadditions vary somewhat, although polar media tend to give higher yields—fluorinated solvents are more effective than their non-fluorinated analogues, and alkoxide or amine bases work better than others.
See also
References
- ↑ Rigby, J. H.; Pigge, F. C. Org. React. 1997, 51, 351. doi:10.1002/0471264180.or051.03
- ↑ Harmata, M.; Elahmad, S.; Barnes, C. L. Tetrahedron Lett. 1995, 36, 1397.
- 1 2 Bingham, R. C.; Dewar, M. J. S.; Lo, D. H. J. Am. Chem. Soc. 1975, 97, 1302.
- ↑ Henning, R.; Hoffmann, H. M. R. Tetrahedron Lett. 1982, 23, 2305.
- ↑ Hill, A. E.; Hoffmann, H. M. R. J. Am. Chem. Soc. 1974, 96, 4597.
- 1 2 Hoffmann, H. M. R. Angew. Chem. Int. Ed. Engl. 1984, 23, 1.
- ↑ Takaya, H.; Makino, S.; Hayakawa, Y.; Noyori, R. J. Am. Chem. Soc. 1978, 100, 1765.
- ↑ Giguere, R. J.; Rawson, D. I.; Hoffmann, H. M. R. Synthesis 1978, 902.
- ↑ Mann, J.; Wilde, P. D.; Finch, M. W. J. Chem. Soc., Chem. Commun. 1985, 1543.
- ↑ West, F. G.; Hartke-Karger, C.; Koch, D. J.; Kuehn, C. E.; Arif, A. M. J. Org. Chem. 1993, 58, 6795.
- ↑ Harmata, M.; Elomari, S.; Barnes, C. L. J. Am. Chem. Soc. 1996, 118, 2860.
- ↑ White, J. D.; Fukuyama, Y. J. Am. Chem. Soc. 1979, 101, 226.
- ↑ Sato, T.; Watanabe, M.; Noyori, R. Tetrahedron Lett. 1978, 4403.
- ↑ Marshall, J. A.; Anderson, N. H.; Johnson, P. C. J. Org. Chem. 1970, 35, 186.
- ↑ Huffman, M. A.; Liebeskind, L. S. J. Am. Chem. Soc. 1993, 115, 4895.