A value

A-Values are numerical values used in the determination of the most stable orientation of atoms in a molecule (Conformational Analysis), as well as a general representation of steric bulk. A-values are derived from energy measurements of a monosubstituted cyclohexane ring.[1] Substituents on a cyclohexane ring prefer to reside in the equatorial position to the axial. The difference in Gibbs free energy (ΔG) between the higher energy conformation (axial substitution) and the lower energy conformation (equatorial substitution) is the A-value for that particular substituent.
Utility
A-values help predict the conformation of cyclohexane rings. The most stable conformation will be the one which has the substituent or substituents equatorial. When multiple substituents are taken into consideration, the conformation where the substituent with the largest A-value is equatorial is favored.

The utility of A-values can be generalized for use outside of cyclohexane conformations. A-values can help predict the steric effect of a substituent. In general, the larger a substituent’s A-value, the larger the steric effect of that substituent. Methyl has an A-value of 1.74 while tert-butyl has an A-value of ~5. Because the A-value of tert-butyl is higher, tert-butyl has a larger steric effect than methyl. This difference in steric effects can be used to help predict reactivity in chemical reactions.
Free energy considerations
Steric effects play a major role in the assignment of configurations in cyclohexanes. One can use steric hindrances to determine the propensity of a substituent to reside in the axial or equatorial plane. It is known that axial bonds are more hindered than the corresponding equatorial bonds. This is because substituents in the axial position are relatively close to two other axial substituents. This makes it very crowded when bulky substituents are oriented in the axial position. These types of steric interactions are commonly known as 1,3 diaxial interactions.[2] These types of interactions are not present with substituents at the equatorial position.
There are generally considered three principle contributions to the conformational free energy:[3]
- Bayer strain, defined as the strain arising from deformation of bond angles.
- Pitzer strain, defined as the torsional strain arising from 1,2 interactions between groups attached to contiguous carbons,
- Van der Waals interactions, which are similar to 1,3 diaxial interactions.
Enthalpic components
When comparing relative stability, 6- and 7-atom interactions can be used to approximate differences in enthalpy between conformations. Each 6-atom interaction is worth 0.9 kcal/mol and each 7-atom interaction is worth 4 kcal/mol.[4]
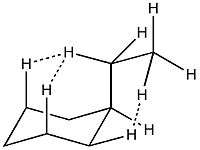

Entropic components
Entropy also plays a role in a substituent’s preference for the equatorial position. The entropic component is determined by the following formula:

Where σ is equal to the number of micro states available for each conformation.

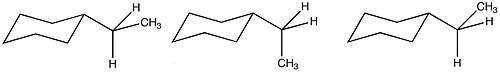
Due to the larger number of possible conformations of ethyl cyclohexane, the A value is reduced from what would be predicted based purely on enthalpic terms. Due to these favorable entropic conditions, the steric relevance of an ethyl group is similar to that of a methyl substituent.
Table of A-Values
The following table lists some common A-values in kcal/mol:[5][6][7][8]
| Substituent | A-Value | Substituent | A-Value | Substituent | A-Value |
|---|---|---|---|---|---|
| D | 0.006 | CH2Br | 1.79 | OSi(CH3)3 | 0.74 |
| F | 0.15 | CH(CH3)2 | 2.15 | OH | 0.87 |
| Cl | 0.43 | c-C6H11 | 2.15 | OCH3 | 0.6 |
| Br | 0.38 | C(CH3)3 | >4 | OCD3 | 0.56 |
| I | 0.43 | Ph | 3 | OCH2CH3 | 0.9 |
| CN | 0.17 | C2H | 1.35 | O-Ac | 0.6 |
| NC | 0.21 | CO2− | 1.92 | O-TFA | 0.68 |
| NCO | 0.51 | CO2CH3 | 1.27 | OCHO | 0.27 |
| NCS | 0.28 | CO2Et | 1.2 | O-Ts | 0.5 |
| N=C=NR | 1 | CO2iPr | 0.96 | ONO2 | 0.59 |
| CH3 | 1.7 | COCl | 1.25 | NH2 | 1.6 |
| CF3 | 2.1 | COCH3 | 1.17 | NHCH3 | 1 |
| CH2CH3 | 1.75 | SH | 0.9 | N(CH3)3 | 2.1 |
| CH=CH2 | 1.35 | SMe | 0.7 | NH3+ | 1.9 |
| CCH | 0.41 | SPh | 0.8 | NO2 | 1.1 |
| CH2tBu | 2 | S− | 1.3 | HgBr | ~0 |
| CH2OTs | 1.75 | SOPh | 1.9 | HgCl | 0.3 |
| SO2Ph | 2.5 | Si(CH3)3 | 2.5 |
These values have been measured at different institutes under different conditions.
Applications
Predicting reactivity
One of the original experiments performed by Winston and Holness was measuring the rate of oxidation in trans and cis substituted rings using a Chromium catalyst. The large tBu group used locks the conformation of each molecule placing it equatorial (cis compound shown).

It was observed that the cis compound underwent oxidation at a much faster rate than the trans compound. The proposition was that the large hydroxyl group in the axial position was disfavored and formed the carbonyl more readily to relieve this strain. The trans compound had rates identical to those found in the monosubstituted cyclohexanol.

Approximating intramolecular force strength using A-Values
Using the A-Values of the hydroxyl and isopropyl subunit, the energetic value of a favorable intramolecular hydrogen bond can be calculated.[9]
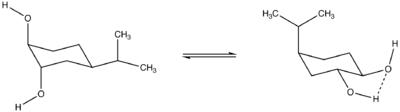
Limitations
A-Values are measured using a mono-substituted cyclohexane ring, and are an indication of only the sterics a particular substituent imparts on the molecule. This leads to a problem when there are possible stabilizing electronic factors in a different system. The carboxylic acid substituent shown below is axial in the ground state, despite a positive A-Value. From this observation, it is clear that there are other possible electronic interactions that stabilize the axial conformation.

Other considerations
It is important to note that A-values do not predict the physical size of a molecule, only the steric effect. For example, the tert-butyl group (A-value=4.9) has a larger A-value than the trimethylsilyl group (A-value=2.5), yet the tert-butyl group actually occupies less space. This difference can be attributed to the longer length of the carbon-silicon bond as compared to the carbon-carbon bond of the tert-butyl group. The longer bond allows for less interactions with neighboring substituents, which effectively makes the trimethylsilyl group less sterically hindering, thus, lowering its A-value.[2] This can also be seen when comparing the halogens. Bromine, iodine, and chlorine all have similar A-values even though their atomic radii differ.[4] A-values then, predict the apparent size of a substituent, and the relative apparent sizes determine the differences in steric effects between compounds. Thus, A-values are useful tools in determining compound reactivity in chemical reactions.
References
- ↑ Muller, P (1994). "Glossary of terms used in physical organic chemistry (IUPAC Recommendations 1994)". Pure and Applied Chemistry. 66 (5): 1077–1184. doi:10.1351/pac199466051077.
- 1 2 Hoffman, Robert V. (2004). Organic Chemistry [An Intermediate Text] (second ed.). New Jersey: John Wiley and Sons, Inc. p. 167. ISBN 978-0-471-45024-5.
- ↑ Anderson, J. Edgar (1974). Dynamic Chemistry [Topics in Current Chemistry]. Springer-Verlag. p. 139. doi:10.1007/3-540-06471-0.
- 1 2 Anslyn, Eric V.; Dougherty, Dennis A. (2006). Modern Physical Organic Chemistry. Sausalito, CA: University Science Books. pp. 104–105. ISBN 978-1-891389-31-3.
- ↑ E.L. Eliel, S.H. Wilen and L.N. Mander, Stereochemistry of Organic Compounds, Wiley, New York (1994). ISBN 81-224-0570-3
- ↑ Eliel, E.L.; Allinger, N.L.; Angyal, S.J.; G.A., Morrison (1965). Conformational Analysis. New York: Interscience Publishers.
- ↑ Hirsch, J.A. (1967). Topics in Stereochemistry (first ed.). New York: John Wiley & Sons,Inc. p. 199.
- ↑ Romers, C.; Altona, C.; Buys, H.R.; Havinga, E. (1969). Topics in Stereochemistry (fourth ed.). New York: John Wiley & Sons,Inc. p. 40.
- ↑ Huang, C.-Y.; Cabell, L.A.; Anslyn, E.V. (1994). "Molecular Recognition of Cyclitols by Neutral Polyaza-Hydrogen-Bonding Receptors: The Strength and Influence of Intramolecular Hydrogen Bonds between Vicinal Alcohols". Journal of the American Chemical Society. 116 (7): 2778–2792. doi:10.1021/ja00086a011.