Aarskog–Scott syndrome
| Aarskog–Scott syndrome | |
|---|---|
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q87.1 |
| ICD-9-CM | 759.89 |
| OMIM | 100050 |
| DiseasesDB | 29329 |
| MedlinePlus | 001654 |
| MeSH | C535331 |
| Orphanet | 915 |
Aarskog–Scott syndrome is a rare disease inherited as autosomal dominant or X-linked and characterized by short stature, facial abnormalities, skeletal and genital anomalies.
The Aarskog–Scott syndrome (AAS) is also known as the Aarskog syndrome, faciodigitogenital syndrome, shawl scrotum syndrome and faciogenital dysplasia.
Signs and symptoms
The Aarskog–Scott syndrome is a disorder with short stature, hypertelorism, downslanting palpebral fissures, anteverted nostrils, joint laxity, shawl scrotum, and developmental delays. The physical phenotype varies with age and postpuberal males may have only minor remnant manifestations of the prepuberal phenotype.
- Growth
- mild to moderate short stature evident by 1–3 years of age
- delayed adolescent growth spurt
- Performance
- slight (dull normal) to moderate mental deficiency
- hyperactivity and attention deficit
- social performance usually good
- Face
- rounded face
- widow's peak hairline
- wide-set eyes (hypertelorism)
- droopy eyelids (blepharoptosis)
- downslanting eye slits (palpebral fissures)
- small nose with nostrils tipped forward (anteverted)
- underdeveloped mid-portion of the face (maxilla)
- wide groove above the upper lip (broad philtrum)
- crease below the lower lip
- delayed eruption of teeth
- top portion (upper helix) of the ear folded over slightly
- Hands and feet
- small, broad hands and feet
- short fingers and toes (brachydactyly)
- in-curving of the 5th finger (clinodactyly)
- mild interdigital webbing, between fingers as well as toes
- single transverse "simian crease" in palm
- broad thumbs and big toes
- Neck
- short neck
- webbing of sides of the neck
- Chest
- mild pectus excavatum (sunken chest)
- Abdomen
- protruding navel
- inguinal hernias
- Genitalia
Genetics
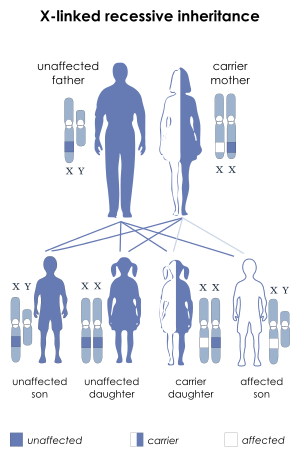
Aarskog–Scott syndrome is transmitted in an X-linked recessive manner. The sons of female carriers are at 50% risk of being affected with the syndrome. The daughters of female carriers are at 50% risk of being carriers themselves. Females may have mild manifestations of the syndrome. The syndrome is caused by mutation in a gene called FGD1 in band p11.21 on the X chromosome.
Pathophysiology
The Aarskog–Scott syndrome is due to mutation in the FGD1 gene. FGD1 encodes a guanine nucleotide exchange factor (GEF) that specifically activates Cdc42, a member of the Rho (Ras homology) family of the p21 GTPases. By activating Cdc42, FGD1 protein stimulates fibroblasts to form filopodia, cytoskeletal elements involved in cellular signaling, adhesion, and migration. Through Cdc42, FGD1 protein also activates the c-Jun N-terminal kinase (JNK) signaling cascade, a pathway that regulates cell growth, apoptosis, and cellular differentiation.
Within the developing mouse skeleton, FGD1 protein is expressed in precartilaginous mesenchymal condensations, the perichondrium and periosteum, proliferating chondrocytes, and osteoblasts. These results suggest that FGD1 signaling may play a role in the biology of several different skeletal cell types including mesenchymal prechondrocytes, chondrocytes, and osteoblasts. The characterization of the spatiotemporal pattern of FGD1 expression in mouse embryos has provided important clues to the understanding of the pathogenesis of Aarskog–Scott syndrome.
It appears likely that the primary defect in Aarskog–Scott syndrome is an abnormality of FGD1/Cdc42 signaling resulting in anomalous embryonic development and abnormal endochondral and intramembranous bone formation.
Diagnosis
Genetic testing may be available for mutations in the FGDY1 gene. Genetic counseling is indicated for individuals or families who may carry this condition, as there are overlapping features with fetal alcohol syndrome.[1]
Treatment
Similar to all genetic diseases Aarskog–Scott syndrome cannot be cured, although numerous treatments exist to increase the quality of life.[2]
Surgery may be required to correct some of the anomalies, and orthodontic treatment may be used to correct some of the facial abnormalities. Trials of growth hormone have been effective to treat short stature in this disorder.[3]
Prognosis
Mild degrees of mental slowness may be present, but affected children usually have good social skills. Some males may exhibit reduced fertility.
Some recent findings have included cystic changes in the brain and generalized seizures . There may be difficulty growing in the first year of life in up to one-third of cases. Misaligned teeth may require orthodontic correction. An undescended testicle will require surgery.
Adenylosuccinate lyase deficiency (MIM 103050, ADSL) is a rare autosomal recessive disease causing severe mental retardation and/or autistic features.1,2 Seizures are often observed (80%),3 varying in age of onset (from newborn to late childhood) and nature (tonic-clonic, “suppression burst” pattern, West syndrome, etc.), and are very often resistant to all medication. Around 50% of the children show autistic-like behaviour.4 Microcephaly is rare (1/13 of reported cases). Non-specific anomalies of the brain, such as hypoplasia of the vermis, cerebral atrophy,5 lack of myelination,6 white matter anomalies,7 and lissencephaly4 have often been described.
Other Complications: • Low self-esteem • Social difficulties related to physical problems • Male infertility in those with both testes undescended • Problems with the structure of the heart • Accumulation of fluid in tissues of body (lymphedema, cystic hygroma) • Failure to thrive in infants.
History
The syndrome is named for Dagfinn Aarskog, a Norwegian pediatrician and human geneticist who first described it in 1970,[4] and for Charles I. Scott, Jr., an American medical geneticist who independently described the syndrome in 1971.[5]
References
- ↑ "Fetal Alcohol Syndrome: Guidelines for Referral and Diagnosis" (PDF). Centers for Disease Control and Prevention. 2004.
- ↑ "Aarskog Syndrome (AAS)". DoveMed. 2014. Retrieved 18 June 2014.
- ↑ Darendeliler F.; Larsson P.; Neyzi O.; et al. (October–November 2003). "Growth hormone treatment in Aarskog syndrome: analysis of the KIGS (Pharmacia International Growth Database) data". J. Pediatr. Endocrinol. Metab. 16 (8): 1137–42. doi:10.1515/jpem.2003.16.8.1137. PMID 14594174.
- ↑ Aarskog D. (1970). "A familial syndrome of short stature associated with facial dysplasia and genital anomalies". J. Pediatr. 77 (5): 856–61. doi:10.1016/S0022-3476(70)80247-5. PMID 5504078.
- ↑ Scott CI (1971). "Unusual facies, joint hypermobility, genital anomaly and short stature: a new dysmorphic syndrome". Birth Defects Orig. Artic. Ser. 7 (6): 240–6. PMID 5173168.
- Jones, Kenneth Lyons (2005). Smith's Recognizable Patterns of Human Malformation (6th ed.). Philadelphia: WB Saunders. ISBN 0-7216-2359-X.
- Orrico A.; Galli L.; Cavaliere ML.; et al. (2004). "Phenotypic and molecular characterisation of the Aarskog–Scott syndrome: a survey of the clinical variability in light of FGD1 mutation analysis in 46 patients". Eur. J. Hum. Genet. 12 (1): 16–23. doi:10.1038/sj.ejhg.5201081. PMID 14560308.
External links
- Aarskog syndrome (Aarskog–Scott syndrome), detailed up-to-date information in OMIM (PediaBook.com)
- Faciogenital Dysplasia (Aarskog–Scott syndrome), detailed up-to-date information in OMIM (Online Mendelian Inheritance in Man)
- Images of clinical features of Aarskog–Scott syndrome
- Pediatric Database Pedbase
- synd/343 at Who Named It?