Benson group increment theory
Benson Group Increment Theory (BGIT), or Group Increment Theory, uses the experimentally calculated heat of formation for individual groups of atoms to calculate the entire heat of formation for a molecule under investigation. This can be a quick and convenient way to determine theoretical heats of formation without conducting tedious experiments. The technique was developed by Professor Sidney William Benson of the University of Southern California.
Heats of formations are intimately related to bond dissociation energies and thus are important in understanding chemical structure and reactivity.[1] Furthermore, although the theory is old, it still is practically useful as one of the best group additivity methods aside from computational methods such as molecular mechanics. However, the BGIT has its limitations, and thus cannot always predict the precise heat of formation.
Origin
Benson and Buss originated the BGIT in a 1958 paper.[2] Within this manuscript, Benson and Buss proposed four approximations:
- A Limiting Law for Additivity Rules.
- Zero-Order Approximation. Additivity of Atomic Properties
- First Order Approximation. Additivity of Bond Properties
- Second Order Approximation. Additivity of Group Properties.
These approximations account for the atomic, bond, and group contributions to heat capacity (Cp), enthalpy (ΔH°), and entropy (ΔS°). The most important of these approximations to the group increment theory is the Second Order Approximation, because this approximation "leads to the direct method of writing the properties of a compound as the sum of the properties of its group."[3]
The Second Order Approximation accounts for two molecular atoms or structural elements that are within relative proximity to one another (approximately 3-5 Angstroms as proposed in the paper). By using a series of disproportionation reactions of symmetrical and asymmetrical framework, Benson and Buss concluded that neighboring atoms within the disproportionation reaction understudy are not affected by the change. In the symmetrical reaction the cleavage between the CH2 in both reactants leads to one product formation. Though difficult to see, one can see that the neighboring carbons are not changed as the rearrangement occurs. In the asymmetrical reaction the hydroxyl-methyl bond is cleaved and rearranged on the ethyl moiety of the methoxyethane. Clearly the methoxy and hydroxyl rearrangement display clear evidence that the neighboring groups are not affected in the disproportionation reaction.
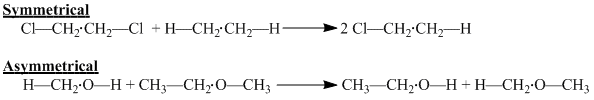
The "disproportionation" reactions that Benson and Buss refer to are termed loosely as "radical disproportionation" reactions.[4] From this they termed a "group" as a polyvalent atom connected together with its ligands. However, they noted that under all approximations ringed systems and unsaturated centers do not follow additivity rules due to their preservation under disproprotionation reactions. One can understand this as you must break a ring at more than one site to actually undergo a disproportionation reaction. This holds true with double and triple bonds, as you must break them multiple times to break their structure. They concluded that these atoms must be considered as distinct entities. Hence we see Cd and CB groups which take into account these groups as being individual entities. Furthermore, this leaves error for ring strain as we will see in its limitations.

From this Benson and Buss concluded that the ΔHf of any saturated hydrocarbon can be precisely calculated due to the only two groups being a methylene [C-(C)2(H)2] and the terminating methyl group [C-(C)(H)3].[5] Benson later began to compile actual functional groups from the Second Order Approximation.[6][7] Ansylyn and Dougherty explained in simple terms how the group increments, or Benson increments, are derived from experimental calculations.[8] By calculating the ΔΔHf between extended saturated alkyl chains (which is just the difference between two ΔHf values), as shown in figure 1 to the right, one can approximate the value of the C-(C)2(H)2 group by averaging the ΔΔHf's. Once this is determined, all one needs to do is take the total value of ΔHf subtract the ΔHf caused by the C-(C)2(H)2 group(s), and then divide that number by two (due to two C-(C)(H)3 groups) and you now have the value of the C-(C)(H)3 group. From the knowledge of these two groups, Benson moved forward obtain and list functional groups derived from countless numbers of experimentation from many sources, some of which are displayed below.
Applications

As stated above, BGIT can be used to calculate heats of formation which are important in understanding the strengths of bonds and entire molecules. Furthermore, the method can be used to quickly estimate whether a reaction is endothermic or exothermic. These values are for gas phase thermodynamics and typically at 298K. Benson and coworkers have continued collecting data since their 1958 publication and have since published even more group increments including strained rings, radicals, halogens, and more.[7][9][10][11] Even though BGIT was introduced in 1958 and would seem to be antiquated in the modern age of advanced computing, the theory still finds practical applications. In a 2006 paper, Gronert states: "Aside from molecular mechanics computer packages, the best known additivity scheme is Benson's."[12] Fishtik and Datta also give credit to BGIT, "Despite their empirical character, GA methods continue to remain a powerful and relatively accurate technique for the estimation of thermodynamic properties of the chemical species, even in the era of supercomputers"[13]
When calculating the heat of formation, all the atoms in the molecule must be accounted for (hydrogen atoms are not included as specific groups). Figure 1 displays a simple application for predicting the standard enthalpy of isobutylbenzene.
First, it is usually very helpful to start off by numbering the atoms. It is much easier than to list the specific groups along with the corresponding number from Table 2. Each individual atom is accounted for where CB-(H) accounts for one benzene carbon a hydrogen atom. This would be multiplied by five, since there are five CB-(H) atoms. The CB-(C) molecule further accounts for the other benzene carbon attached to the butyl group. The C-(CB)(C)(H)2 accounts for the carbon linked to the benzene group on the butyl moiety. The 2' carbon of the butyl group would be C-(C)3(H) because it is a tertiary carbon (connecting to three other carbon atoms). The final calculation comes from the CH3 groups connected to the 2' carbon; C-(C)(H)3. The total calculations add to -5.15kcal/mol (-21.6 kJ/mole), which is identical to the experimental value which can be found in the National Institute of Standards and Technology Chemistry WebBook [14]

Figure 2 gives an example from the literature in which the BGIT was used to corroborate experimental evidence of the enthalpy of formation of benzo[k]fluoranthene.[15] The experimental value was determined to be 296.6 kJ/mole with a standard deviation of 6.4 kJ/mole. This is within the error of the BGIT and is in good agreement with the calculated value. Notice that the carbons at the fused rings are treated differently than regular benzene carbons.[7] Not only can the BGIT be used to confirm experimental values but can also be used to confirm theoretical values.
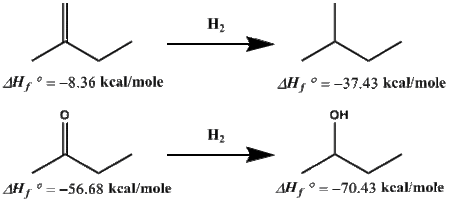
Another way to use BGIT is shown in Figure 3 where the thermodynamics of a simplified hydrogenation reaction is compared between an alkene (2-methyl-1-butene) and a ketone(2-butanone). This is a thermodynamic argument and kinetics are ignored. As determined by the enthalpy's below the corresponding molecules, the enthalpy of reaction for 2-methyl-1-butene going to 2-methyl-butane is -29.07 kcal/mole which is in great agreement with the value calculated from NIST,[14] -28.31 kcal/mole. For 2-butanone going to 2-butanol, enthalpy of reaction is -13.75 kcal/mole, which again is in excellent agreement with -14.02 kcal/mole. While both reactions are thermodynamically favored, the alkene will be far more exothermic than the corresponding ketone.
Limitations
As powerful as it is, BGIT does have several limitations which restrict its usage.
Inaccuracy
There is an overall 2-3 Kcal/mol error using the Benson Group Increment Theory to calculate the △Hf. The value of each group is estimated on the base of the average △△Hf0shown above and there will be a dispersion around the average △△Hf0. Also, it can only be as accurate as the experimental accuracy. That's the derivation of the error and there is nearly no way to make it more accurate.
Group Availability
The BGIT is based on empirical data and heat of formation. Some groups are too hard to measure, so not all the existing groups are available in the Table above.Sometimes approximation should be made when we meet those unavailable groups. For example, we need to approximate C as Ct and N as NI in C≡N,which clearly cause more inaccuracy, which is another drawback.
Ring Strain, Intermolecular and Intramolecular Interactions
In the BGIT, we assumed that a CH2 always makes a constant contribution to △Hf0 for a molecule. However, a small ring such as cyclobutane leads to a substantial failure for the BGIT, because of its strain energy. A series of correction terms for common ring systems has been developed, with the goal of obtaining accurate △Hf0 values for cyclic system. Representative values are given in the Table shown below. Note that these are not identically equal to the accepted strain energies for the parent ring system, although they are quite close. The group increment correction for a cyclobutane is based on △Hf0 values for a number of structures, and represents an average value that gives the best agreement with the range of experimental data. In contrast, the strain energy of cyclobutane is specific to the parent compound, with their new corrections, it is now possible to predict △Hf0 values for strained ring system, by first adding up all the basic group increments and then adding appropriate ring strain correction values.
The same as ring system, corrections have been made to other situations such as Gauche alkane with a 0.8kcal/mol correction, cis alkene with a 1.0kcal/mol correction, respectively.
Also, the BGIT fails when conjugation and interactions between functional groups exist,[16][17] such as intermolecular and intramolecular hydrogen bonding shown in the right figure, which limits its accuracy and usage in a few cases.
References
- ↑ Benson, S. W. Journal of Chemical Education 1965, 42, 502-518
- ↑ Sidney W. Benson and Jerry H. Buss (September 1958). "Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties". J. Chem. Phys. 29 (3): 546. doi:10.1063/1.1744539.
- ↑ Benson, S. W.; Buss, J. H. Journal of Chemical Physics 1958, 29, 546-572.
- ↑ International Union of Pure and Applied Chemistry. "disproportionation". Compendium of Chemical Terminology (Accessed December 03, 2008)
- ↑ Souders, M.; Matthews, C. S.; Hurd C. O., Ind. & Eng. Chemistry 1949, 41, 1037-1048.
- ↑ Benson, S. W.; Cruicksh, F. R.; Golden, D. M., et al. Chemical Reviews 1969, 69, 279-324.
- 1 2 3 Benson, S. W.; Cohen, N. Chemical Reviews 1993, 93, 2419-2438.
- ↑ Eric V. Anslyn and Dennis A. Dougherty Modern Physical Organic Chemistry University Science Books, 2006.
- ↑ S. W. Benson, Thermochemical Kinetics: Methods for the Estimation of Thermochemical Data and Rate Parameters 2d ed., John Wiley & Sons, New York, 1973.
- ↑ E. S. Domalski, E. D. Hearing; "Estimation of the Thermodynamic Properties of C-H-N-O-S-X Compounds at 298K", J. Phys. Chem. Ref. Data, Vol. 22, 805-1159 (1993)
- ↑ N. Cohen; "Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of C-H and C-H-O Compounds", J. Phys. Chem. Ref. Data, Vol. 25, 1411-1481 (1996)
- ↑ Gronert, S. J. Org. Chem.2006,71,1209-1219.
- ↑ Fishtik, I.; Datta, R. J. Phys. Chem. A 2003, 107, 6698-6707.
- 1 2 National Institute of Standards and Technology- (accessed December 3, 2008).
- ↑ Diogo, H.P.; Piedade, M.E.M. J. Chem. Thermodynamics 2002,34, 173-184.
- ↑ Knoll, H. Schliebs, R.; Scherzer, H. Reaction Kinetics and Catalysis Letters 1978, 8, 469-475.
- ↑ Sudlow, K.; Woolf, A.A. Journal of Fluorine Chemistry 1995, 71, 31-37.