Protecting group
A protecting group or protective group is introduced into a molecule by chemical modification of a functional group to obtain chemoselectivity in a subsequent chemical reaction. It plays an important role in multistep organic synthesis.
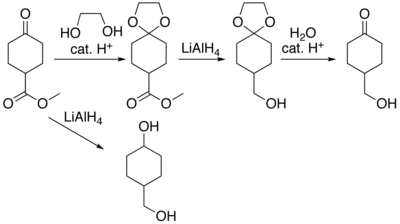
In many preparations of delicate organic compounds, some specific parts of their molecules cannot survive the required reagents or chemical environments. Then, these parts, or groups, must be protected. For example, lithium aluminium hydride is a highly reactive but useful reagent capable of reducing esters to alcohols. It will always react with carbonyl groups, and this cannot be discouraged by any means. When a reduction of an ester is required in the presence of a carbonyl, the attack of the hydride on the carbonyl has to be prevented. For example, the carbonyl is converted into an acetal, which does not react with hydrides. The acetal is then called a protecting group for the carbonyl. After the step involving the hydride is complete, the acetal is removed (by reacting it with an aqueous acid), giving back the original carbonyl. This step is called deprotection.
Protecting groups are more commonly used in small-scale laboratory work and initial development than in industrial production processes because their use adds additional steps and material costs to the process. However, the availability of a cheap chiral building block can overcome these additional costs (e.g. shikimic acid for oseltamivir).
Common protecting groups
Alcohol protecting groups
Protection of alcohols:
- Acetyl (Ac) – Removed by acid or base (see Acetoxy group).
- Benzoyl (Bz) – Removed by acid or base, more stable than Ac group.
- Benzyl (Bn) – Removed by hydrogenolysis. Bn group is widely used in sugar and nucleoside chemistry.
- β-Methoxyethoxymethyl ether (MEM) – Removed by acid.
- Dimethoxytrityl, [bis-(4-methoxyphenyl)phenylmethyl] (DMT) – Removed by weak acid. DMT group is widely used for protection of 5'-hydroxy group in nucleosides, particularly in oligonucleotide synthesis.
- Methoxymethyl ether (MOM) – Removed by acid.
- Methoxytrityl [(4-methoxyphenyl)diphenylmethyl] (MMT) – Removed by acid and hydrogenolysis.
- p-Methoxybenzyl ether (PMB) – Removed by acid, hydrogenolysis, or oxidation.
- Methylthiomethyl ether – Removed by acid.
- Pivaloyl (Piv) – Removed by acid, base or reductant agents. It is substantially more stable than other acyl protecting groups.
- Tetrahydropyranyl (THP) – Removed by acid.
- Tetrahydrofuran (THF) - Removed by acid.
- Trityl (triphenylmethyl, Tr) – Removed by acid and hydrogenolysis.
- Silyl ether (most popular ones include trimethylsilyl (TMS), tert-butyldimethylsilyl (TBDMS), tri-iso-propylsilyloxymethyl (TOM), and triisopropylsilyl (TIPS) ethers) – Removed by acid or fluoride ion. (such as NaF, TBAF (tetra-n-butylammonium fluoride, HF-Py, or HF-NEt3)). TBDMS and TOM groups are used for protection of 2'-hydroxy function in nucleosides, particularly in oligonucleotide synthesis.
- Methyl Ethers – Cleavage is by TMSI in dichloromethane or acetonitrile or chloroform. An alternative method to cleave methyl ethers is BBr3 in DCM
- Ethoxyethyl ethers (EE) – Cleavage more trivial than simple ethers e.g. 1N hydrochloric acid[1]
Amine protecting groups
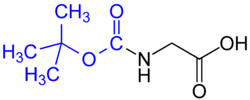
Protection of amines:
- Carbobenzyloxy (Cbz) group – Removed by hydrogenolysis
- p-Methoxybenzyl carbonyl (Moz or MeOZ) group – Removed by hydrogenolysis, more labile than Cbz
- tert-Butyloxycarbonyl (BOC) group (common in solid phase peptide synthesis) – Removed by concentrated strong acid (such as HCl or CF3COOH), or by heating to >80 °C.
- 9-Fluorenylmethyloxycarbonyl (FMOC) group (Common in solid phase peptide synthesis) – Removed by base, such as piperidine
- Acetyl (Ac) group is common in oligonucleotide synthesis for protection of N4 in cytosine and N6 in adenine nucleic bases and is removed by treatment with a base, most often, with aqueous or gaseous ammonia or methylamine. Ac is too stable to be readily removed from aliphatic amides.
- Benzoyl (Bz) group is common in oligonucleotide synthesis for protection of N4 in cytosine and N6 in adenine nucleic bases and is removed by treatment with a base, most often with aqueous or gaseous ammonia or methylamine. Bz is too stable to be readily removed from aliphatic amides.
- Benzyl (Bn) group – Removed by hydrogenolysis
- Carbamate group – Removed by acid and mild heating.
- p-Methoxybenzyl (PMB) – Removed by hydrogenolysis, more labile than benzyl
- 3,4-Dimethoxybenzyl (DMPM) – Removed by hydrogenolysis, more labile than p-methoxybenzyl
- p-methoxyphenyl (PMP) group – Removed by ammonium cerium(IV) nitrate (CAN)
- Tosyl (Ts) group – Removed by concentrated acid (HBr, H2SO4) & strong reducing agents (sodium in liquid ammonia or sodium naphthalenide)
- Troc (trichloroethyl chloroformate ) group – Removed by Zn insertion in the presence of acetic acid
- Other Sulfonamides (Nosyl & Nps) groups – Removed by samarium iodide, tributyltin hydride[2]
Carbonyl protecting groups
Protection of carbonyl groups:
- Acetals and Ketals – Removed by acid. Normally, the cleavage of acyclic acetals is easier than of cyclic acetals.
- Acylals – Removed by Lewis acids.
- Dithianes – Removed by metal salts or oxidizing agents.
Carboxylic acid protecting groups
Protection of carboxylic acids:
- Methyl esters – Removed by acid or base.
- Benzyl esters – Removed by hydrogenolysis.
- tert-Butyl esters – Removed by acid, base and some reductants.
- Esters of 2,6-disubstituted phenols (e.g. 2,6-dimethylphenol, 2,6-diisopropylphenol, 2,6-di-tert-butylphenol) – Removed at room temperature by DBU-catalyzed methanolysis under high-pressure conditions.[3]
- Silyl esters – Removed by acid, base and organometallic reagents.
- Orthoesters – Removed by mild aqueous acid to form ester, which is removed according to ester properties.
- Oxazoline – Removed by strong hot acid (pH < 1, T > 100 °C) or alkali (pH > 12, T > 100 °C), but not e.g. LiAlH4, organolithium reagents or Grignard (organomagnesium) reagents
Phosphate protecting groups
- 2-cyanoethyl – removed by mild base. The group is widely used in oligonucleotide synthesis.
- Methyl (Me) – removed by strong nucleophiles e.c. thiophenole/TEA.
Terminal alkyne protecting groups
- Propargyl alcohols in the Favorskii reaction,
- Silyl groups, especially in protection of the acetylene itself.[4]
Orthogonal protection
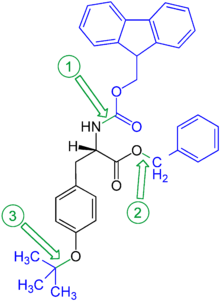
Orthogonal protection is a strategy allowing the deprotection of multiple protective groups one at a time each with a dedicated set of reaction conditions without affecting the other. In the example shown, the protected amino acid tyrosine, the benzyl ester can be removed by hydrogenolysis, the fluorenylmethylenoxy group (Fmoc) by bases (such as piperidine), and the phenolic tert-butyl ether cleaved with acids (e.g. with trifluoroacetic acid).
A common example for this application, the Fmoc-peptide synthesis, in which peptides are grown in solution and on solid phase is very important.[5] The protecting groups in solid-phase synthesis with regard to the reaction conditions such as reaction time, temperature and reagents can be standardized so that they are carried out by a machine, while yields of well over 99% can be achieved. Otherwise, the separation of the resulting mixture of reaction products is virtually impossible.[6]
The technique was introduced in the field of peptide synthesis by Robert Bruce Merrifield in 1977.[7] As a proof of concept orthogonal deprotection is demonstrated in a photochemical transesterification by trimethylsilyldiazomethane utilizing the kinetic isotope effect:[8]
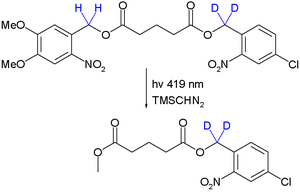
Due to this effect the quantum yield for deprotection of the right-side ester group is reduced and it stays intact. Significantly by placing the deuterium atoms next to the left-side ester group or by changing the wavelength to 254 nm the other monoarene is obtained.
Criticism
In a 2007 paper[9] Phil Baran notes that even though the textbooks state that the use of protective groups is unavoidable and that they are ideally easily added and removed, in practical terms in organic synthesis their use adds two synthetic steps (protection-deprotection sequence) to a chemical sequence and sometimes dramatically lowers chemical yield. Crucially, added complexity impedes the use of synthetic total synthesis in drug discovery. In contrast biomimetic synthesis does not employ protective groups. As an alternative, Baran presented a novel protective-group free synthesis of the compound hapalindole U. The previously published synthesis[10][11][12] according to Baran, contained 20 steps with multiple protective group manipulations (two confirmed):
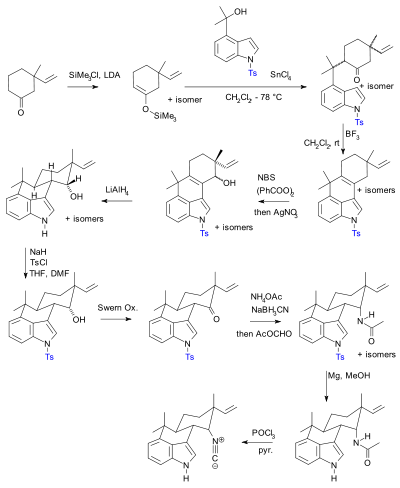 Hideaki Muratake's 1990 synthesis using Tosyl protecting groups (shown in blue). |
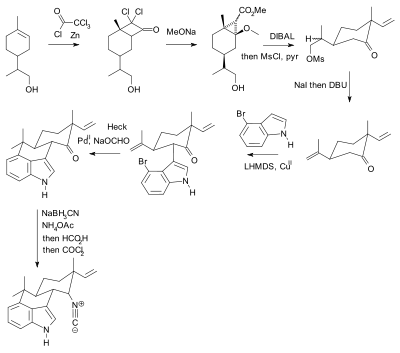 Phil Baran's protecting-group free synthesis, reported in 2007. |
Industrial applications
Although the use of protecting groups is not preferred in industrial syntheses, they are still used in industrial contexts, e.g.:
- Oseltamivir (Tamiflu, an antiviral drug) synthesis by Roche
- Sucralose (sweetener)
External links
For introduction of protecting group and mechanism of deprotection See : http://www.biocis.u-psud.fr/spip.php?article332
- Senior undergraduate study notes on this subject, from Prof. Rizzo.
- A further set of study notes in tutorial form, with guidance and comments, from Profs. Grossman and Cammers.
- A review by Prof. Kocienski.
- A user site excerpting the classic Greene and Wuts text regarding stability of a few key groups, from this reference's extensive tables.
- protecting group from organic-reaction.com
References
- ↑ Kamaya, Yasushi; T Higuchi (2006). "Metabolism of 3,4-dimethoxycinnamyl alcohol and derivatives by Coriolus versicolor". FEMS Microbiology Letters. 24 (2–3): 225–229. doi:10.1111/j.1574-6968.1984.tb01309.x.
- ↑ Moussa, Ziad; D. Romo (2006). "Mild deprotection of primary N-(p-toluenesufonyl) amides with SmI2 following trifluoroacetylation". Synlett. 2006 (19): 3294–3298. doi:10.1055/s-2006-951530.
- ↑ Romanski, J.; Nowak, P.; Kosinski, K.; Jurczak, J. (Sep 2012). "High-pressure transesterification of sterically hindered esters". Tetrahedron Lett. 53 (39): 5287–5289. doi:10.1016/j.tetlet.2012.07.094.
- ↑ Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2000). Organic Chemistry. Oxford University Press. p. 1291. ISBN 978-0198503460.
- ↑ Chan, Weng C.; White, Peter D. (2004). Fmoc Solid Phase Peptide Synthesis. Oxford University Press. ISBN 0-19-963724-5.
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis, S. 10–12.
- ↑ Merrifield, R. B.; Barany, G.; Cosand, W. L.; Engelhard, M.; Mojsov, S. (1977). "Proceedings of the 5th American Peptide Symposium". Biochemical Education. 7 (4): 93–94. doi:10.1016/0307-4412(79)90078-5.
- ↑ Blanc, Aurélien; Bochet, Christian G. (2007). "Isotope Effects in Photochemistry: Application to Chromatic Orthogonality". Org. Lett. 9 (14): 2649–2651. doi:10.1021/ol070820h.
- ↑ Baran, Phil S.; Maimone, Thomas J.; Richter, Jeremy M. (22 March 2007). "Total synthesis of marine natural products without using protecting groups". Nature (446): 404–408. doi:10.1038/nature05569.
- ↑ Synthetic studies of marine alkaloids hapalindoles. Part I Total synthesis of (±)-hapalindoles J and M Tetrahedron, Volume 46, Issue 18, 1990, Pages 6331–6342 Hideaki Muratake and Mitsutaka Natsume doi:10.1016/S0040-4020(01)96005-3
- ↑ Synthetic studies of marine alkaloids hapalindoles. Part 2. Lithium aluminum hydride reduction of the electron-rich carbon-carbon double bond conjugated with the indole nucleus Tetrahedron, Volume 46, Issue 18, 1990, Pages 6343–6350 Hideaki Muratake and Mitsutaka Natsume doi:10.1016/S0040-4020(01)96006-5
- ↑ Synthetic studies of marine alkaloids hapalindoles. Part 3 Total synthesis of (±)-hapalindoles H and U Tetrahedron, Volume 46, Issue 18, 1990, Pages 6351–6360 Hideaki Muratake, Harumi Kumagami and Mitsutaka Natsume doi:10.1016/S0040-4020(01)96007-7
External links
| Wikiquote has quotations related to: Protecting group |