GeNMR

GeNMR method (GEnerate NMR structures) is the first fully automated template-based method of protein structure determination that utilizes both NMR chemical shifts and NOE -based distance restraints.[1]
In addition to the template-based approach, the GeNMR webserver also offers an ab initio protein folding mode that starts folding from an extended structure. The GeNMR web server produces an ensemble of PDB coordinates within a period ranging from 20 minutes to 4 hours, depending on protein size, server load, quality and type of experimental information, and selected protocol options. GeNMR webserver is composed of two parts, a front-end web-interface (written in Perl and HTML) and a back-end consisting of eight different alignment, structure generation and structure optimization programs along with three local databases.
GeNMR input
GeNMR accepts and processes backbone and side chain 1H, 13C or 15N chemical shift data of almost any combination (HA only, HN only, HA+HN only, HA+HN+sidechain H, CA only, CA+CB only, CA+CO only, HA+CA+CB, HN+CA+CB, HN+15N only, HN,+15N+CA, HN+15N+CA+CB, etc.). This allows GeNMR to handle small peptides (where only H shifts are typically measured) to large proteins (where only N or C shifts might be available). The input files must include chemical shift data in NMR-STAR 2.1 format and distance restraints in XPLOR/CNS format (see more info here). The minimum sequence length is 30 residues.
GeNMR output
The output for a typical GeNMR structure calculation consists of a user-defined set of lowest energy PDB coordinates in a simple, downloadable text format. In addition, details about the overall energy score (prior to and following energy minimization) and chemical shift correlations (between the observed and calculated shifts) is provided at the top of the output page. If score failed to decrease below a certain threshold, a warning is printed at the top of the page.
GeNMR sub-programs
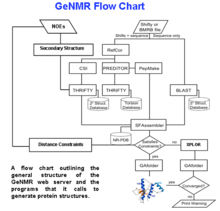
A flow chart describing the processing logic used in GeNMR is shown on the right. GeNMR makes use of a number of well-known programs and databases. These include Proteus2 to perform structural modeling, PREDITOR to calculate torsion angles from chemical shifts, PPT-DB for comparative modeling and alignment and CS23D to calculate protein structures from chemical shifts only. GeNMR also uses several well-known external programs, including Rosetta for ab initio folding without NOEs and XPLOR-NIH for NOE-based simulated annealing and refinement. A more complete list of GeNMR sub-programs is listed on the CS23D page.
GeNMR homology modelling
GeNMR uses homology modeling and sequence/structure threading to rapidly generate a first-pass model of the query protein. The use of homology modeling/threading in GeNMR allows a considerable speed-up in its structure calculations since homology models can often be generated and refined in a minute or two.
GeNMR genetic algorithm
GeNMR also makes use of genetic algorithms to allow configurational sampling and structural refinement using non-differentiable scores, such as ShiftX chemical shift scores. GeNMR's genetic algorithm creates a population of initial structures and then uses combinations of mutations, cross-overs, segment swaps and writhe movements to comprehensively sample conformation space. The 25 lowest energy structures are then selected, duplicated and carried to the next round of conformational sampling.
GeNMR scoring functions
The potential functions used in GeNMR are derived from those used in CS23D and Proteus2. The knowledge-based potentials include information on predicted/known secondary structure, radius of gyration, hydrogen bond energies, number of hydrogen bonds, allowed backbone and side chain torsion angles, atom contact radii (bump checks), disulfide bonding information and a modified threading energy based on the Bryant and Lawrence potential. The chemical shift component of the GeNMR potential uses weighted correlation coefficients calculated between the observed and SHIFTX calculated shifts of the structure being refined.
GeNMR calculation scenarios
There are six different kinds of calculation scenarios that GeNMR can currently accommodate. These scenarios include:
- chemical shift only—query has homologue in database;
- chemical shift only—query has no homologue in database;
- NOE only—query has homologue in database;
- NOE only—query has no homologue in database;
- NOE and chemical shift—query has homologue in database;
- NOE and chemical shift—query has no homologue in database.
See also
- Chemical Shift
- NMR
- Nuclear magnetic resonance spectroscopy
- Protein nuclear magnetic resonance spectroscopy
- Protein dynamics#Domains and protein flexibility
- Protein
- Random Coil Index
- CS23D
- Protein Chemical Shift Re-Referencing
- Protein secondary structure
- Protein Chemical Shift Prediction
- Chemical shift index
- Protein NMR
- ShiftX
- Protein structure prediction
References
- ↑ Berjanskii, Mark; Tang P; Liang J; Cruz JA; Zhou J; Zhou Y; Bassett E; MacDonell C; Lu P; Lin G; Wishart DS. (April 30, 2009). "GeNMR: a web server for rapid NMR-based protein structure determination.". Nucleic Acids Res. 37: W670–7. doi:10.1093/nar/gkp280. PMC 2703936
 . PMID 19406927.
. PMID 19406927.