Local-density approximation
Local-density approximations (LDA) are a class of approximations to the exchange–correlation (XC) energy functional in density functional theory (DFT) that depend solely upon the value of the electronic density at each point in space (and not, for example, derivatives of the density or the Kohn–Sham orbitals). Many approaches can yield local approximations to the XC energy. However, overwhelmingly successful local approximations are those that have been derived from the homogeneous electron gas (HEG) model. In this regard, LDA is generally synonymous with functionals based on the HEG approximation, which are then applied to realistic systems (molecules and solids).
In general, for a spin-unpolarized system, a local-density approximation for the exchange-correlation energy is written as
![E_{xc}^{\mathrm{LDA}}[\rho] = \int \rho(\mathbf{r})\epsilon_{xc}(\rho)\ \mathrm{d}\mathbf{r}\ ,](../I/m/82784592c3830dbc02f552efeac6056d34f99252.svg)
where ρ is the electronic density and εxc is the exchange-correlation energy per particle of a homogeneous electron gas of charge density ρ. The exchange-correlation energy is decomposed into exchange and correlation terms linearly,

so that separate expressions for Ex and Ec are sought. The exchange term takes on a simple analytic form for the HEG. Only limiting expressions for the correlation density are known exactly, leading to numerous different approximations for εc.
Local-density approximations are important in the construction of more sophisticated approximations to the exchange-correlation energy, such as generalized gradient approximations or hybrid functionals, as a desirable property of any approximate exchange-correlation functional is that it reproduce the exact results of the HEG for non-varying densities. As such, LDA's are often an explicit component of such functionals.
Applications
Local density approximations, as with Generalised Gradient Approximations (GGA) are employed extensively by solid state physicists in ab-initio DFT studies to interpret electronic and magnetic interactions in semiconductor materials including semiconducting oxides and Spintronics. The importance of these computational studies stems from the system complexities which bring about high sensitivity to synthesis parameters necessitating first-principles based analysis. The prediction of Fermi level and band structure in doped semiconducting oxides is often carried out using LDA incorporated into simulation packages such as CASTEP and DMol3.[1] However an underestimation in Band gap values often associated with LDA and GGA approximations may lead to false predictions of impurity mediated conductivity and/or carrier mediated magnetism in such systems.[2]
Homogeneous electron gas
Approximation for εxc depending only upon the density can be developed in numerous ways. The most successful approach is based on the homogeneous electron gas. This is constructed by placing N interacting electrons in to a volume, V, with a positive background charge keeping the system neutral. N and V are then taken to infinity in the manner that keeps the density (ρ = N / V) finite. This is a useful approximation as the total energy consists of contributions only from the kinetic energy and exchange-correlation energy, and that the wavefunction is expressible in terms of planewaves. In particular, for a constant density ρ, the exchange energy density is proportional to ρ⅓.
Exchange functional
The exchange-energy density of a HEG is known analytically. The LDA for exchange employs this expression under the approximation that the exchange-energy in a system where the density is not homogeneous, is obtained by applying the HEG results pointwise, yielding the expression[3][4]
![E_{x}^{\mathrm{LDA}}[\rho] = - \frac{3}{4}\left( \frac{3}{\pi} \right)^{1/3}\int\rho(\mathbf{r})^{4/3}\ \mathrm{d}\mathbf{r}\ .](../I/m/13db139ef06d3f00ce7908d54e35e4c9995ee804.svg)
Correlation functional
Analytic expressions for the correlation energy of the HEG are available in the high- and low-density limits corresponding to infinitely-weak and infinitely-strong correlation. For a HEG with density ρ, the high-density limit of the correlation energy density is[3]

and the low limit

where the Wigner-Seitz radius is related to the density as

The analytical expression for the full range of densities has been proposed based on the many-body perturbation theory. The error as compared to the near-exact quantum Monte Carlo simulation is on the order of milli-Hartree.
- Chachiyo's correlation functional: [5]

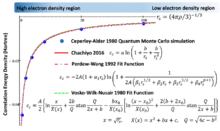
Accurate quantum Monte Carlo simulations for the energy of the HEG have been performed for several intermediate values of the density, in turn providing accurate values of the correlation energy density.[6] The most popular LDA's to the correlation energy density interpolate these accurate values obtained from simulation while reproducing the exactly known limiting behavior. Various approaches, using different analytic forms for εc, have generated several LDA's for the correlation functional, including
Predating these, and even the formal foundations of DFT itself, is the Wigner correlation functional obtained perturbatively from the HEG model.[11]
Spin polarization
The extension of density functionals to spin-polarized systems is straightforward for exchange, where the exact spin-scaling is known, but for correlation further approximations must be employed. A spin polarized system in DFT employs two spin-densities, ρα and ρβ with ρ = ρα + ρβ, and the form of the local-spin-density approximation (LSDA) is
![E_{xc}^{\mathrm{LSDA}}[\rho_{\alpha},\rho_{\beta}] = \int\mathrm{d}\mathbf{r}\ \rho(\mathbf{r})\epsilon_{xc}(\rho_{\alpha},\rho_{\beta})\ .](../I/m/b6a755c41ca0ad2fb7789b3236c4df47eb3c221d.svg)
For the exchange energy, the exact result (not just for local density approximations) is known in terms of the spin-unpolarized functional:[12]
![E_{x}[\rho_{\alpha},\rho_{\beta}] = \frac{1}{2}\bigg( E_{x}[2\rho_{\alpha}] + E_{x}[2\rho_{\beta}] \bigg)\ .](../I/m/fc8b4f4641da8817988c144cfa4782c1bf0aea4e.svg)
The spin-dependence of the correlation energy density is approached by introducing the relative spin-polarization:

corresponds to the paramagnetic spin-unpolarized situation with equal and spin densities whereas corresponds to the ferromagnetic situation where one spin density vanishes. The spin correlation energy density for a given values of the total density and relative polarization, εc(ρ,ς), is constructed so to interpolate the extreme values. Several forms have been developed in conjunction with LDA correlation functionals.[7][13]




Exchange-correlation potential
The exchange-correlation potential corresponding to the exchange-correlation energy for a local density approximation is given by[3]

In finite systems, the LDA potential decays asymptotically with an exponential form. This is in error; the true exchange-correlation potential decays much slower in a Coulombic manner. The artificially rapid decay manifests itself in the number of Kohn–Sham orbitals the potential can bind (that is, how many orbitals have energy less than zero). The LDA potential can not support a Rydberg series and those states it does bind are too high in energy. This results in the HOMO energy being too high in energy, so that any predictions for the ionization potential based on Koopman's theorem are poor. Further, the LDA provides a poor description of electron-rich species such as anions where it is often unable to bind an additional electron, erroneously predicating species to be unstable.[8][14]
References
- ↑ Segall, M.D.; Lindan, P.J (2002). "First-principles simulation: ideas, illustrations and the CASTEP code". Journal of Physics: Condensed Matter. 14 (11): 2717. Bibcode:2002JPCM...14.2717S. doi:10.1088/0953-8984/14/11/301.
- ↑ Assadi, M.H.N; et al. (2013). "Theoretical study on copper's energetics and magnetism in TiO2 polymorphs" (PDF). Journal of Applied Physics. 113 (23): 233913. arXiv:1304.1854
 . Bibcode:2013JAP...113w3913A. doi:10.1063/1.4811539.
. Bibcode:2013JAP...113w3913A. doi:10.1063/1.4811539. - 1 2 3 Parr, Robert G; Yang, Weitao (1994). Density-Functional Theory of Atoms and Molecules. Oxford: Oxford University Press. ISBN 978-0-19-509276-9.
- ↑ Dirac, P. A. M. (1930). "Note on exchange phenomena in the Thomas-Fermi atom". Proc. Cambridge Phil. Roy. Soc. 26 (3): 376–385. Bibcode:1930PCPS...26..376D. doi:10.1017/S0305004100016108.
- ↑ Teepanis Chachiyo (2016). "Simple and accurate uniform electron gas correlation energy for the full range of densities". J. Chem. Phys. 145 (2): 021101. doi:10.1063/1.4958669.
- ↑ D. M. Ceperley and B. J. Alder (1980). "Ground State of the Electron Gas by a Stochastic Method". Phys. Rev. Lett. 45 (7): 566–569. Bibcode:1980PhRvL..45..566C. doi:10.1103/PhysRevLett.45.566.
- 1 2 S. H. Vosko, L. Wilk and M. Nusair (1980). "Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis". Can. J. Phys. 58 (8): 1200. Bibcode:1980CaJPh..58.1200V. doi:10.1139/p80-159.
- 1 2 J. P. Perdew and A. Zunger (1981). "Self-interaction correction to density-functional approximations for many-electron systems". Phys. Rev. B. 23 (10): 5048. Bibcode:1981PhRvB..23.5048P. doi:10.1103/PhysRevB.23.5048.
- ↑ L. A. Cole and J. P. Perdew (1982). "Calculated electron affinities of the elements". Phys. Rev. A. 25 (3): 1265. Bibcode:1982PhRvA..25.1265C. doi:10.1103/PhysRevA.25.1265.
- ↑ John P. Perdew and Yue Wang (1992). "Accurate and simple analytic representation of the electron-gas correlation energy". Phys. Rev. B. 45 (23): 13244–13249. Bibcode:1992PhRvB..4513244P. doi:10.1103/PhysRevB.45.13244.
- ↑ E. Wigner (1934). "On the Interaction of Electrons in Metals" (abstract). Phys. Rev. 46 (11): 1002–1011. Bibcode:1934PhRv...46.1002W. doi:10.1103/PhysRev.46.1002.
- ↑ Oliver, G. L.; Perdew, J. P. (1979). "Spin-density gradient expansion for the kinetic energy". Phys. Rev. A. 20 (2): 397–403. Bibcode:1979PhRvA..20..397O. doi:10.1103/PhysRevA.20.397.
- ↑ von Barth, U.; Hedin, L. (1972). "A local exchange-correlation potential for the spin polarized case". J. Phys. C: Solid State Phys. 5 (13): 1629–1642. Bibcode:1972JPhC....5.1629V. doi:10.1088/0022-3719/5/13/012.
- ↑ Fiolhais, Carlos; Nogueira, Fernando; Marques Miguel (2003). A Primer in Density Functional Theory. Springer. p. 60. ISBN 978-3-540-03083-6.