Stereoelectronic effect
The stereoelectronic effect is the effect on molecular structures, physical properties and reactivities due to the molecules' electronic structures, in particular the interaction between atomic and/or molecular orbitals.[1] Typical stereoelectronic effects with specific orbital overlaps generally lead to a specific molecular conformation or energy differentiation among various transition states that would lead to a particular reaction selectivity.
The stereoelectronic effect, along with the steric effect, inductive effect, mesomeric effect, etc., is one of the key theories in illustrating unusual selectivity, reactivity and stability cases in the course of organic chemistry. Its application has widely spread in organic methodology and organic synthesis. This topic is now entering biochemistry and pharmaceutical chemistry.
Stereoelectronic effect generally includes a donor–acceptor interaction. The donor is usually a higher bonding or nonbonding orbital and the acceptor is often a low-lying antibonding orbital as shown in the scheme below. As known from the orbital–orbital interaction requirement, if this stereoelectronic effect is to be favored, the donor–acceptor orbitals must have a low energy gap and they must retain antiperiplanar geometry to allow for perfect interacting direction.
Stereoelectronic effect contains a large variety of subtopics, including anomeric effects and hyperconjugation.[2]
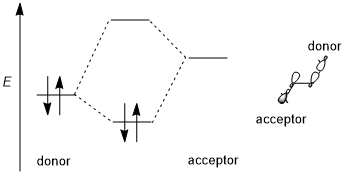
Trend of different orbitals
Take the simplest CH2X–CH3 system as an example; the donor orbital is σ(C–H) orbital and the acceptor is σ*(C–X). When moving from fluorine to chlorine, then to bromine, the electronegativity of the halogen and the energy level of the σ*(C–X) orbitals decreases.[3] Consequently, the general trend of acceptors can be summarized as: π*(C=O)>σ*(C–Hal)>σ*(C–O)>σ*(C–N)>σ*(C–C), σ*(C–H). For donating orbitals, the nonbonding orbitals, or the lone pairs, are generally more effective than bonding orbitals due to the high energy levels. Also, different from acceptors, donor orbitals require less polarized bonds. Thus, the general trends for donor orbitals would be: n(N)>n(O)>σ(C–C), σ(C–H)>σ(C–N)>σ(C–O)>σ(C–S)>σ(C–Hal).[4]

Stereoelectronic effect can be directional in specific cases. The radius of sulfur is much larger than the radius of carbon and oxygen. Thus the differences in C–S bond distances generate a much amplified difference in the two stereoelectronic effects in 1,3-dithiane (σ(C–H) → σ*(C–S)) than in 1,3-dioxane(σ(C–H) → σ*(C–O)).[3] The differences between C–C and C–S bonds shown below causes a significant difference in the distances between C–S and two C–H bonds. The shorter the difference is, the better the interaction and the stronger the stereoelectronic effect.[3]

Influence on stability
If there is an electropositive substituent (e.g. –SiR3, –SnR3, –HgR, etc.) at the β-position of carbocation, the positive charge could be stabilized which is also due largely to the stereoelectronic effect (illustrated below using –SiR3 as an example). The orientation of the two interacting orbitals can have a significant effect on the stabilization effect (σ(C–Si) → empty p orbital), where antiperiplanar (180°) > perpendicular (90°) > syn (0°).[5]

Influence on conformation
Gauche effect
One structural consequence of acyclic systems due to the stereoelectronic effect is the gauche effect.[6] In 1,2-difluoroethane, despite the steric clash, the preferred conformation is the gauche one because σ(C–H) is a good donor and σ*(C–F) is a good acceptor and the stereoelectronic effect (σ(C–H) → σ*(C–F)) requires the energy minimum to be gauche instead of anti.[7]

This gauche effect has a profound effect in bio-chemical research. In (2S,4R)-4-hydroxyproline fragment, the gauche interaction favors one the conformer that can bind selectively to the active site of pVHL, a domain in collagen, one of the most abundant animal protein structures and can lead to proteasomal degradation of HIF-α subunit.[8]

Special effects of fluorine substituent
The stereoelectronic effect can have a significant influence in pharmaceutical research. Generally, the substitution of hydrogen by fluorine could be regarded as a way to tune both the hydrophobicity and the metabolism rate. It has a profound influence on conformations. In anisole, the methyl group prefers the coplanar geometry with the phenyl group by about 3.0 kcal/mol, while the trifluoromethoxybenzene favors the O–CF3 bond to be perpendicular to the phenyl group. One explanation is the enhanced steric size of trifluoromethyl group compared to methyl group; yet, more importantly, once the trifluomethyl group rotates outside the phenyl plane, which orienting the C–F bond antiperiplanar (with the [C(aryl)–C(aryl)–O–C(F3)] dihedral angle in the energy minimized structure being around 90°) to the nonbonding orbital (the lone pair) on the oxygen atom which can benefit more of the stereoelectronic effect (n(O) → σ*(C–F)).[9]

Further studies illustrate that even for only one or two hydrogen atoms in a methyl group being replaced by a fluorine atom, the distortion in the structure can also be significant, with the [C(aryl)–C(aryl)–O–C(H2F)] dihedral angle in the energy minimized structure being around 24° and the [C(aryl)–C(aryl)–O–C(HF2)] dihedral angle 33°.[9]
Influence on reaction selectivity
Reductive cyclizations
Although the energy difference between coplanar anisole and its isomer is quite large, the rotation between the O–CH3 bond becomes favorable when the electronic properties of methoxy group on aromatic rings need to be altered to stabilize an unusual intermediate or a transition state. In the following reaction, the regioselectivity could be rationalized as the out-of-plane rotation of the O–C bond which changes the methoxy group from an in-plane donor group to an out-of-plane acceptor group.[10]
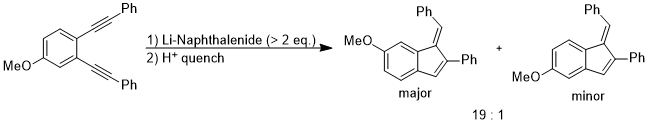
The intermediate of the above reaction is the di-anion and the stereoelectronic effect that stabilizes this intermediate over the other one is the fact that the anionic charge at the para position could delocalize to the oxygen atom via orbital interaction: π(benzene) → σ*(O–CH3).[10]

Hydrogenation
The other substituents on the benzene ring can also affect the electron density on the aromatic ring and in turn influence the selectivity. In the hydrogenation of ketones using CBS catalysts, the ketone coordinate to the boron atom with the lone pair on the oxygen atom. In the following example, the substituent's influence can be passed to differentiate the two lone pairs on the oxygen atom.[11]
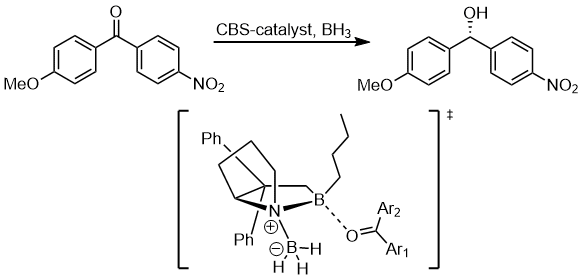
The stereoelectronic interaction in the starting material is the n(O) → σ*(Ccarbonyl–Caryl). The electron-withdrawing substituent on the benzene ring depletes the electron density on the aromatic ring and thus makes the σ*(Ccarbonyl–Caryl(nnitrile)) orbital a better acceptor than σ*(Ccarbonyl–Caryl(methoxy)) and these two stereoelectronic interactions would use different lone pairs on the oxygen atom. Also, the better the stereoelectronic interaction is, the less reactive it is to coordinate to the boron atom. This would result in the intermediate to coordinate boron atom with the oxygen lone pair syn to the nitrile-aryl group.[11]
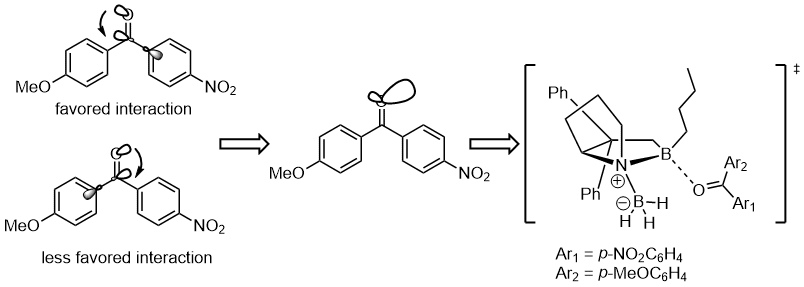
Influence on thermodynamics
Influence on equilibrium
The stereoelectronic effect influences the thermodynamics of equilibrium and even between different resonance structures. For example, the following equilibrium could be achieved via a cascade of pericyclic reactions.
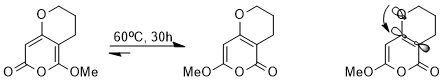
Though nearly every step is reversible, one of the two structures is strongly favored. With an oxygen atom in the ring, the double bonds cannot resonate with each other and are localized. As the σ*(C(sp2)–C(sp2)) orbital is a better acceptor than σ*(C(sp3)–C(sp3)), the stereoelectronic interaction of n(O) → σ*(C(sp2)–C(sp2)) has a much stronger stabilizing effect than n(O) → σ*(C(sp3)–C(sp3)); and thus the structure with better stereoelectronic interaction is favored thermodynamically in equilibrium.[12]
Another example of the preference in the equilibrium within the area of pericyclic reaction is shown below. The stereoelectronic effects that affect the quilibrium is the interaction between the delocalized “banana bonds” and the empty p orbital on the boron atom.[13]

Influence on resonance structures
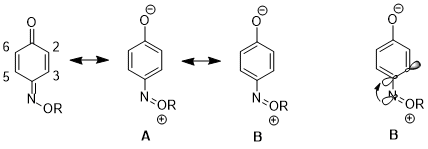
In another case, the stereoelectronic effect can result in a preference for one resonance structure over the other which leads to further consequences in reactivity. For 1,4-benzoquinone monoxime, significant differences separate the physical properties and reactivities between C2-C3 double bond and C5-C6 double bond with the J23 higher than J56.[14] The C2-C3 double bond is more isolated and tends not to undergo Diels–Alder reaction with ethylene.[15] This illustrates a preference of resonance structure B against structure A. The reason for the preference in 1,4-benzoquinone monoxime is the fact that the σ*(C(sp2)–C(sp2)) orbital is a better acceptor in single bonds than in double bonds. The stereoelectronic interaction of n(N) → σ*(C(sp2)–C(sp2)-single bond) is more favorable than n(N) → σ*(C(sp2)–C(sp2)-double bond).[16]
Application in asymmetric Diels–Alder reactions
In the asymmetric Diels–Alder reactions, instead of using chiral ligands or chiral auxiliaries to differentiate the side selectivity of the dienolphiles, the differentiation of face selectivity of the dienes (especially for cyclopentadiene derivatives) using stereoelectronic effects have been reported by Woodward since 1955.[17] A systematic research of facial selectivity using substituted cyclopentadiene or permethylcyclopentadiene derivatives have been conducted and the results can be listed as below.[18]

The stereoelectronic effect affecting the outcome of the facial selectivity of the diene in Diels–Alder reaction is the interaction between the σ(C(sp2)–CH3) (when σ(C(sp2)–X) is a better acceptor than a donor) or σ(C(sp2)–X) (when σ(C(sp2)–X) is a better donor than an acceptor) and the σ* orbital of the forming bond between the diene and the dienophile.[18]

If the two geminal substituents are both aromatic rings with different substituents tuning the electron density, the differentiation of the facial selectivity is also facile where the dienophile approaches to the diene anti to the more electron-rich C–C bond where the stereoelectronic effect in this case is similar to the previous one.[19]

The ring opening of cyclobutene under heating conditions can have two products: inward and outward rotation.
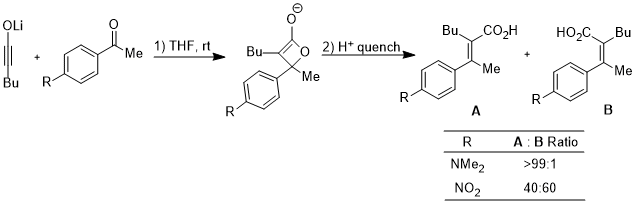
The inward rotation transition state of the second shown below is relatively favored for acceptor R substitutents (e.g. NO2) but is especially disfavored by donor R substituents (e.g. NMe2).[20]
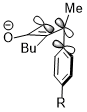
Stereoelectronic effect versus steric clash
Sometimes, stereoelectronic effects can win over extreme steric clash. In a similar cyclobutene ring-opening reaction, the trimethylsilyl group, which is very bulky, still favors the inward rotation. The stereoelectronic effect, which is the interaction shown above when the acceptor orbital is the σ*(Si–CH3), appears to be a more predominant factor in determining the reaction selectivity against the steric hindrance and even wins over the penalty of the disrupted conjugation system of the product due to steric clash.[21]
Furthermore, the acceptor orbitals are not limited to the antibonding orbitals of carbon-heteroatom bonds or the empty orbitals; in the following case, the acceptor orbital is the σ*(B–O) orbital. In the six-membered ring transition state, the stereoelectronic interaction is σ(C–X) → σ*(B–O).[22]

References
- ↑ Cramer, Christopher J. (1996). "Hyperconjugation as it affects conformational analysis". Journal of Molecular Structure: THEOCHEM. 370 (2–3): 135–146. doi:10.1016/S0166-1280(96)04567-8. ISSN 0166-1280.
- ↑ Kirby, A. J. (1983). The Anomeric Effect and Related Stereoelectronic Effects at Oxygen. New York: Springer-Verlag. pp. 3–13.
- 1 2 3 Alabugin, Igor V.; Zeidan, Tarek A. (2002). "Stereoelectronic Effects and General Trends in Hyperconjugative Acceptor Ability of σ Bonds". Journal of the American Chemical Society. 124 (12): 3175–3185. doi:10.1021/ja012633z. ISSN 0002-7863. PMID 11902907.
- ↑ Kirby, A. J. (1996). Stereoelectronic Effect. Oxford: Oxford University Press.
- ↑ Carey, A. F. S. (2007). Advanced organic Chemistry Part A: Structure and Mechanism. New York: Springer.
- ↑ Hanack, M. (1965). Conformational Theory. New York and London: Academic Press.
- ↑ Anslyn, E.V. (2005). Modern Physical Organic Chemistry. Herndon: University Science Books.
- ↑ Fujimori, D.G. (2009). Nat. Chem. Bio. 5: 202. Missing or empty
|title=(help) - 1 2 Müller, K.; Faeh, C.; Diederich, F. (2007). "Fluorine in Pharmaceuticals: Looking Beyond Intuition". Science. 317 (5846): 1881–1886. Bibcode:2007Sci...317.1881M. doi:10.1126/science.1131943. ISSN 0036-8075. PMID 17901324.
- 1 2 Peterson, Paul W.; Shevchenko, Nikolay; Alabugin, Igor V. (2013). ""Stereoelectronic Umpolung": Converting a p-Donor into a σ-Acceptor via Electron Injection and a Conformational Change". Organic Letters. 15 (9): 2238–2241. doi:10.1021/ol400813d. ISSN 1523-7060. PMID 23639080.
- 1 2 Corey, E.J.; Helal, Christopher J. (1995). "Novel electronic effects of remote substituents on the oxazaborolidine-catalyzed enantioselective reduction of ketones". Tetrahedron Letters. 36 (50): 9153–9156. doi:10.1016/0040-4039(95)01961-G. ISSN 0040-4039.
- ↑ Venkataraman, Hemalatha; Cha, Jin K. (1989). "The scope and mechanism of rearrangement of 4,6-dialkoxy-2-pyrones". Tetrahedron Letters. 30 (27): 3509–3512. doi:10.1016/S0040-4039(00)99426-7. ISSN 0040-4039.
- ↑ Gridnev, Ilya D.; Tok, Oleg L.; Gridneva, Natalya A.; Bubnov, Yuri N.; Schreiner, Peter R. (1998). "Synthesis and Dynamic Properties of Cycloheptatrienyl(dipropyl)borane. Equilibrium with 7-Dipropylborylnorcaradiene". Journal of the American Chemical Society. 120 (5): 1034–1043. doi:10.1021/ja9724699. ISSN 0002-7863.
- ↑ Norris, R. K.; Sternhell, S. (1969). Aust. J. Chem. 22: 935. Missing or empty
|title=(help) - ↑ Baldwin, J. E.; Norris, R. K. (1981). "Stereoelectronic control in organic chemistry: addition reactions of some 1,4-benzoquinone 4-(O-methyloximes)". J. Org. Chem. 46: 697–703. doi:10.1021/jo00317a011.
- ↑ Perrin, C. L.; Engler, R. E. (1997). "Origin of Apparent Stereoelectronic Effects in Structure and Reactivity of Benzoquinone Monooximes". J. Org. Chem. 62: 687–692. doi:10.1021/jo961386s.
- ↑ Winstein, S.; Shatavsky, M.; Norton, C.; Woodward, R. B. (1955). "7-NORBORNENYL AND 7-NORBORNYL CATIONS". J. Am. Chem. Soc. 77: 4183–4184. doi:10.1021/ja01620a078.
- 1 2 Mehta, Goverdhan; Uma, R. (2000). "Stereoelectronic Control in Diels−Alder Reaction of Dissymmetric 1,3-Dienes". Accounts of Chemical Research. 33 (5): 278–286. doi:10.1021/ar990123s. ISSN 0001-4842. PMID 10813872.
- ↑ Halterman, R. L.; McCarthy, B. A.; McEvoy, M. A. (1992). "Stereoelectronic control of facial selectivity in the Diels-Alder cycloaddition of sterically unbiased 5,5-diarylcyclopentadienes". J. Org. Chem. 57: 5585–5589. doi:10.1021/jo00047a009.
- ↑ Shindo, M.; Sato, Y.; Shishido, J. (1998). "A highly stereoselective synthesis of tri- and tetrasubstituted olefins via ynolates". Tetrahedron Lett. 39: 4857–4860. doi:10.1016/s0040-4039(98)00921-6.
- ↑ Murakami, M.; Hasegawa, M. (2004). "Synthesis and Thermal Ring Opening oftrans-3,4-Disilylcyclobutene". Angew. Chem. Int. Ed. 43: 4874–4876. doi:10.1002/anie.200460144.
- ↑ Schlapbach, A.; Hoffmann, R. W. (2001). "(E)-α-Sulfonamidocrotylboronates as Reagents for the Stereoselective Homoaldol Synthesis". J. Org. Chem. 66: 323–328. doi:10.1002/1099-0690(200101)2001:2<323::aid-ejoc323>3.0.co;2-a.