Pyramidal alkene
Pyramidal alkenes are alkenes in which the two carbon atoms making up the double bond are not coplanar with their four substituents. This deformation from a trigonal planar geometry to a tetrahedral molecular geometry is the result of angle strain induced in the molecule due to geometric constraints. Pyramidal alkenes only exist in the laboratory but are of interest because much can be learned from them about the nature of chemical bonding.[1]
In cycloheptene (1.1) the cis isomer is an ordinary unstrained molecule, but the heptane ring is too small to accommodate a trans-configured alkene group resulting in strain and twisting of the double bond. The p-orbital misalignment is minimized by a degree of pyramidalization. In the related anti-Bredt molecules. it is not pyrimidalization but twisting that dominates.
 Figure 1. Pyramidal alkenes
Figure 1. Pyramidal alkenes
Pyramidalized cage alkenes also exist where symmetrical bending of the substituents predominates without p-orbital misalignment.
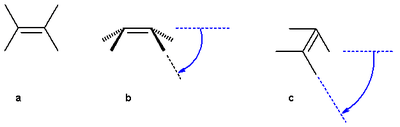 Figure 2. Angle definitions
Figure 2. Angle definitions
The pyramidalization angle φ (b) is defined as the angle between the plane defined by one of the doubly bonded carbons and its two substituents and the extension of the double bond and is calculated as:

the butterfly bending angle or folding angle ψ (c) is defined as the angle between two planes and can be obtained by averaging the two torsional angles R1C=CR3 and R2C=CR4.
In alkenes 1.2 and 1.3 these angles are determined with X-ray crystallography as respectively 32.4°/22.7° and 27.3°/35.6°. Although stable, these alkenes are very reactive compared to ordinary alkenes. They are liable to dimerization to cyclobutadiene compounds or react with oxygen to epoxides.
The compound tetradehydrodianthracene, also with a 35° pyramidalization angle, is synthesized in a photochemical cycloaddition of bromoanthracene followed by elimination of hydrogen bromide.
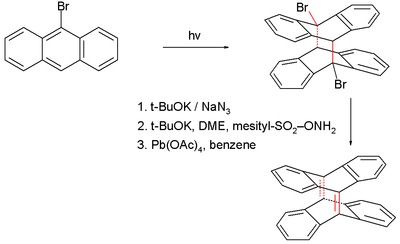 Figure 3. Tetradehydrodianthracene synthesis
Figure 3. Tetradehydrodianthracene synthesis
This compound is very reactive in Diels–Alder reactions due to through-space interactions between the two alkene groups. This enhanced reactivity enabled in turn the synthesis of the first-ever Möbius aromat.
In one study,[2] the strained alkene 4.4 was synthesized with the highest pyramidalizion angles yet, 33.5° and 34.3°. This compound is the double Diels–Alder adduct of the diiodocyclophane 4.1 and anthracene 4.3 by reaction in presence of potassium tert-butoxide in refluxing dibutyl ether through a diaryne intermediate 4.2. This is a stable compound but will slowly react with oxygen to an epoxide when left standing as a chloroform solution.
 Figure 4. Cyclophane anthracene adduct
Figure 4. Cyclophane anthracene adduct
In one study,[3] isolation of a pyramidal alkene is not even possible by matrix isolation at extremely low temperatures unless stabilized by metal coordination:
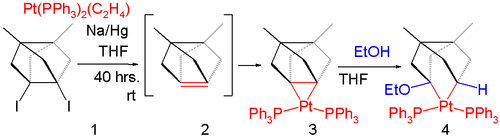 Figure 5. (Ph3P)2Pt complex of 3,7-dimethyltricyclo[3.3.0.03,7]oct-1(5)-ene
Figure 5. (Ph3P)2Pt complex of 3,7-dimethyltricyclo[3.3.0.03,7]oct-1(5)-ene
A reaction of the diiodide 5.1 in Figure 5 with sodium amalgam in the presence of ethylenebis(triphenylphosphine)platinum(0) does not give the intermediate alkene 5.2 but the platinum stabilized 5.3. The sigma bond in this compound is destroyed in reaction with ethanol.
References
- ↑ Vázquez, Santiago; Camps, Pelayo (2005). "Chemistry of pyramidalized alkenes". Tetrahedron. 61 (22): 5147–5208. doi:10.1016/j.tet.2005.03.055.
- ↑ Dolbier, W. R., Jr; Zhai, Y.-A.; Battiste, M. A.; Abboud, K. A.; Ghiviriga, I. (2005). "A Highly Pyramidalized Cage Alkene Formed via the Double Diels-Alder Cycloaddition of syn-4,5,13,14-Bis(dehydro)octafluoroparacyclophane to Anthracene". J. Org. Chem. 70 (25): 10336–10341. doi:10.1021/jo051488v.
- ↑ Theophanous, Fanitsa A.; Tasiopoulos, Anastasios J.; Nicolaides, Athanassios; Zhou, Xin; Johnson, William T. G.; Borden, Weston Thatcher (2006). "Evidence for the Formation of the (Ph3P)2Pt Complex of 3,7-Dimethyltricyclo[3.3.0.03,7]oct-1(5)-ene, the Most Highly Pyramidalized Alkene in a Homologous Series. Isolation and X-ray Structure of the Product of the Ethanol Addition to the Complex". Org. Lett. 8 (14): 3001–3004. doi:10.1021/ol060994j.