Urofacial syndrome
| Ochoa Syndrome | |
|---|---|
| Classification and external resources | |
| OMIM | 236730 |
| DiseasesDB | 32631 |
Ochoa syndrome, also called urofacial syndrome or hydronephrosis with peculiar facial expression,[1] is an autosomal recessive[2] congenital disorder characterized by inverted facial expressions in association with obstructive disease of the urinary tract. The inverted facial expression presented by children with this syndrome allows for early detection of the syndrome, this inverted smile is easy to see when the child is smiling and laughing. Early detection is vital for establishing a better prognosis as urinary related problems associated with this disease can cause harm if left untreated. Incontinence is another easily detectable symptom of the syndrome that is due to detrusor-sphincter discoordination, although it can easily be mistaken for pyelonephritis.
It may be associated with HPSE2.[3]
Characteristics
Infants with the disorder exhibit an inverted smile; they appear to be crying when they are actually smiling, in conjunction with uropathy. They also may be affected by hydronephrosis. Symptoms of this disease can start at very young ages. Many people with this syndrome will die in their teens to early 20s because of the renal failure (uropathy) if not diagnosed and treated. Children with the syndrome have abnormal facial development that cause an inverted smile, nerve connections are however normal. When attempting to smile, the child will appear to cry. Urinary problems arise as a result of a neurogenic bladder. Most patients older than the age of toilet training, present with enuresis, urinary-tract infection, hydronephrosis, and a spectrum of radiological abnormalities typical of obstructive or neurogenic bladders. Radiological abnormalities include things such as: trabeculated bladder, vesicoureteral reflex, external sphincter spasm, pyelonephritis, hyperreflexic bladder, noninhibited detrusor contraction, etc.. Urinary abnormalities might result in renal deterioration and failure. This can be prevented by taking proper measures to restore normal micturition and by taking antibiotics to prevent infections. In some cases, the affected patients become hypertensive and progress to end-stage renal disease, while others become uremic. Additionally, most patients suffer from constipation.[4] Early detection of this syndrome is possible through the peculiar faces that children present.
Cause
Urofacial Syndrome occurs due to either disruption or mutation of a gene on chromosome 10q23q24.[5] The gene is located on a 1 centimorgan interval between D10S1433 and D10S603.[4] Alteration of this gene leads to alteration of facial and urinary developmental fields. This gene is believed to be the HPSE2 gene. The HPSE2 gene is expressed in both the central nervous system as well as the bladder. Heparanase 2 is protein coded by exons 8 and 9 on the HPSE2 gene. This protein is believed to be altered in the case of this syndrome.[6] Studies performed on mice indicate that HPSE2 has no enzymatic activity.[7]
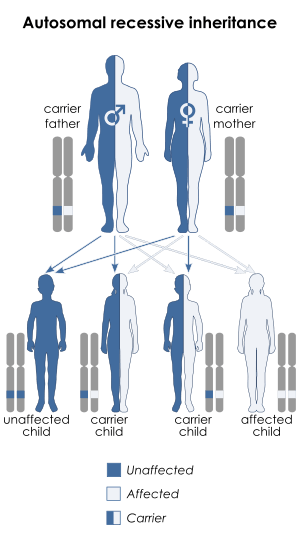
Mutations in the HPSE2 gene on chromosome 10q23q24 have been observed to cause Ochoa Syndrome. This means the defective gene responsible for the disorder is located on an autosome (chromosome 10 is an autosome), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
The relationship between a defective HPSE2 gene and Ochoa syndrome is unclear. There is postulation that the genetic changes may lead to an abnormality in the brain region, evidence for this postulation is that the areas of the brain that control facial expression and urination are in close proximity of each other. Other hypotheses think that the defective heparanase 2 protein may lead to problems with development of the urinary tract or with muscle function in the face and bladder.[8]
Treatment
Treatment can include amoxicillin-clavulanic acid, intravenous fluid administration and paracetamol oral for pain relief.[9] Other treatment varies based on the condition and extent of uropathy.
Epidemiology
Urofacial (Ochoa) syndrome received the Ochoa name because of the first person to describe it back in 1987, Bernardo Ochoa.[4]
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM) 236730
- ↑ Chauve, X.; Missirian, C.; Malzac, P.; Girardot, L.; Guys, J. M.; Louis, C.; Philip, N.; Voelckel, M. A. (Nov 2000). "Genetic homogeneity of the urofacial (Ochoa) syndrome confirmed in a new French family". American Journal of Medical Genetics. 95 (1): 10–12. doi:10.1002/1096-8628(20001106)95:1<10::AID-AJMG3>3.0.CO;2-Z. PMID 11074487.
- ↑ Daly SB, Urquhart JE, Hilton E, et al. (June 2010). "Mutations in HPSE2 Cause Urofacial Syndrome". Am J Hum Genet. 86 (6): 963–969. doi:10.1016/j.ajhg.2010.05.006. PMC 3032078
 . PMID 20560210.
. PMID 20560210. - 1 2 3 Wang, C Y; B Hawkins-Lee; B Ochoa; R D Walker; J X She (June 1997). "Homozygosity and linkage-disequilibrium mapping of the urofacial (Ochoa) syndrome gene to a 1-cM interval on chromosome 10q23-q24.". The American Journal of Human Genetics. 60 (6): 1461–1467. doi:10.1086/515469. PMC 1716147. PMID 9199567.
- ↑ Kulkarni, R; Manish K Arya (15 April 2009). "Ochoa or Urofacial Syndrome". Indian Pediatrics. 1 (1): 445–446. Retrieved November 26, 2012.
- ↑ Daly, Sarah B.; Jill E. Urquhart, Emma Hilton, Edward A McKenzie. Richard A. Kammerer (June 2010). "Mutations in HPSE2 Cause Urofacial Syndrome". AJHG. 86 (6): 963–969. doi:10.1016/j.ajhg.2010.05.006. PMC 3032078. PMID 20560210.
- ↑ Zhang, Yinghui; Denise S. Ryan; Kraig S. Bower; Neta Ilan; Israel Vlodavsky; Gordon W. Laurie (May 2010). "Focus on Molecules: Heparanse". NCBI. 91 (4): 476–7. doi:10.1016/j.exer.2010.05.004. PMC 2933305. PMID 20478306.
- ↑ "Ochoa Syndrome". Genetics Home Reference. Retrieved 30 November 2012.
- ↑ Stamatiou KN, Karakos CD. Urofacial syndrome: A subset of neurogenic bladder dysfunction syndromes?. Indian J Urol 2010;26:582-4