Vitamin B12 total synthesis
The total synthesis of the complex biomolecule vitamin B12 was first accomplished by the collaborating research groups of Robert Burns Woodward at Harvard [1][2][3] and Albert Eschenmoser at ETH [4][5][6][7] in 1972. It is considered a classic in the field of total synthesis of natural products.[8][9][10][11] Work on the synthesis started 1960 at ETH, and in 1961 at Harvard, it was collaboratively pursued after 1965, and required the effort of no less than 91 post-doctoral fellows (mostly at Harvard) and 12 Ph.D. students (at ETH) from 19 different nations.
There are two variants of this synthesis, concomitantly accomplished in 1972. These two syntheses are intricately intertwined chemically, yet they basically differ in their overall strategy of creating the macrocyclic corrin ligand system of the vitamin molecule. The variant collaboratively pursued and finished at Harvard closes the macrocyclic corrin ring between rings A and B (the "A/B variant"), while the synthesis accomplished at ETH achieves the corrin ring closure between rings A and D by a photochemical process (the "A/D variant"). Woodward reported on the A/B variant in lectures published in 1968, [1] 1971,[2] and 1973,[3] culminating in the announcement of the "Total Synthesis of Vitamin B12" in his lecture at the IUPAC Conference in New Delhi, July 1972.[3] Eschenmoser discussed the ETH-contributions to the A/B variant in his Centenary Lecture, published in 1970,[4] and presented the approach to the photochemical A/D variant of the B12 synthesis at the 23rd IUPAC Congress in Boston, published in 1971.[5] A full report on the photochemical variant is given in a Science article [7] of 1977 which is an extended English translation of a 1974 Naturwissenschaften article,[6] based on a lecture given by Eschenmoser at the Chemical Society of Zürich.
The two variants of the chemical synthesis of vitamin B12 have been reviewed by R.V. Stevens [12] and Nicolaou & Sorensen,[8] and discussed in more or less detail in more than 40 other publications.[13][14] The account given here is based on the three published B12 lectures of Woodward [1][2][3] and, therefore, deals with the Harvard-ETH A/B variant only.
The X-ray crystal structure of Vitamin B12 had been determined by Dorothy Hodgkin (Oxford University) in collaboration with Kenneth N. Trueblood (UCLA) and John G. White (Princeton University) in 1956.[15] According to Woodward,[16] an observation made during the synthesis of the Harvard A-D component played an essential role in the conception of the Woodward–Hoffmann rules for orbital symmetry control of organic reactions formulated in 1965.
The molecule
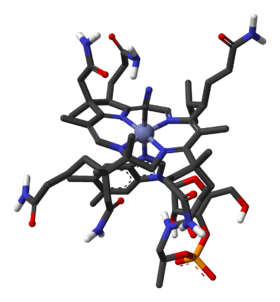
The core of the molecule vitamin B12 (cobalamin) is a corrin structure (depicted in red) with at its center a cobalt ion. Several vitamins exist with different cobalt ligands but the total synthesis concerned the one with a cyano ligand called cyanocobalamin. The corrin rim is lined with methyl groups (8) and lined with amide groups (6) linked through C1 and C2 spacers. A seventh amide group is N-alkylated by a large tail consisting of an isopropanol group, a phosphate group, a ribose group and a dimethylbenzimidazole group. One of the nitrogen atoms on the imidazole is a fifth nitrogen ligand for the cobalt atom. A total of nine carbon atoms on the corrin frame are chiral, adding another challenge to the synthesis.
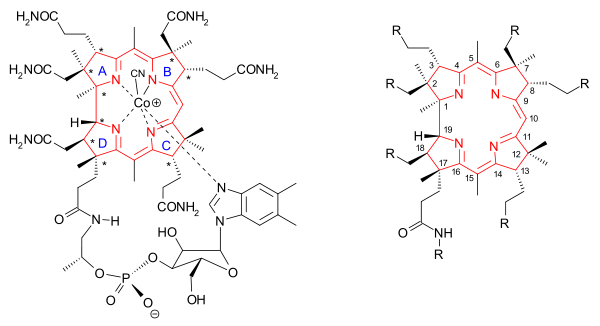 |
| Vitamin B12 overview |
|---|
Retrosynthesis
In retrosynthesis step 1 was easy. It was already established by Bernhauer in 1960 that the tail can be removed from vitamin B12 by amide hydrolysis to cobyric acid and again replaced.[17] The Woodward/Eschenmoser venture is strictly a formal synthesis because the ultimate target was cobyric acid and tail addition was not included.
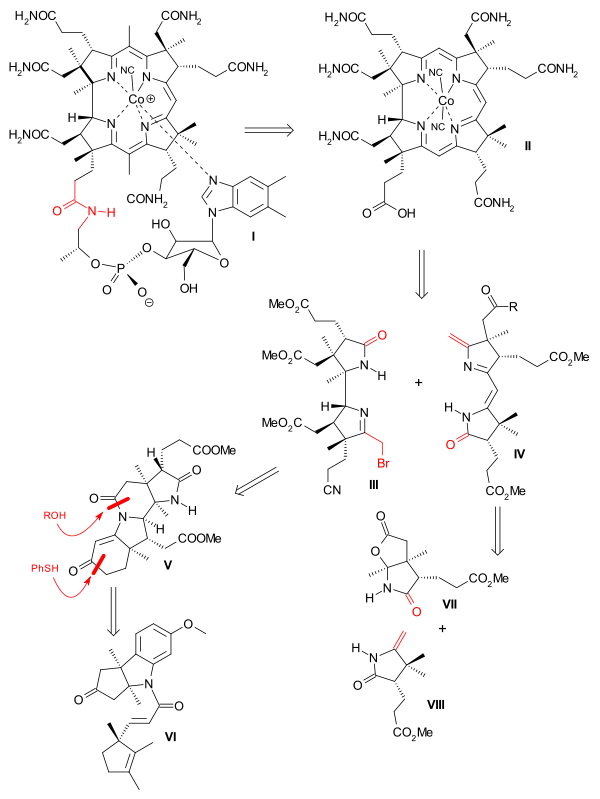 |
| Vitamin B12 retrosynthesis |
|---|
The methyl groups at C5 and C15 were added only after construction of the corrin core. This core was synthesised by joining an AD western part (III) with an BC eastern part (IV). Direct union was not considered feasible due to steric hindrance but both joins were made possible by a sulfur contraction method.
Synthesis
Ring A synthesis
Starting point for the synthesis of ring A was methoxydimethylindol 3 synthesised by condensation of the Schiff base from m-anisidine 1 and acetoin 2. Reaction with the Grignard reagent of propargyl iodide 4 give the propargyl indolenine 5 and ring-closure to 7 was brought about by boron trifluoride and mercuric oxide in methanol through intermediate 6 (electrophilic addition) with the two methyl groups forced into a cis-relationship.
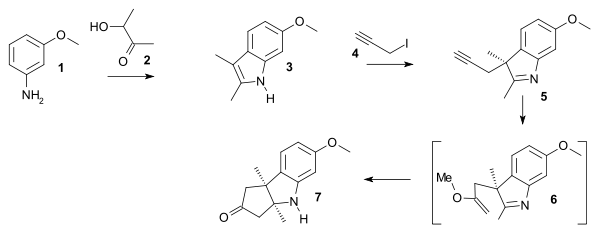 |
| Vitamin B12 AD ring part A |
|---|
This compound existed as a mixture of two enantiomers (racemic) and chiral resolution using (−)-alpha-phenylethylisocyanate was employed for the isolation of the (+)-enantiomer.
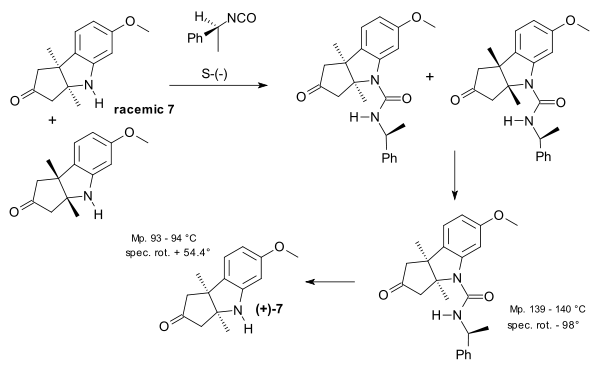 |
| Vitamin B12 ring A chiral resolution |
|---|
Ring D synthesis
The D ring was synthesized starting from chiral (S)-camphor 8 which was converted to oxime 9 (oxidation / hydroxylamine ), then to amide 10 (hydrolysis), lactam 11 (acid-amide condensation), N-nitroso compound 12, diazo compound 13 and to cyclopentene 14 (carbene, methyl group migration). Reduction (LiAlH4) gave alcohol 15, oxidation (chromic acid) gave aldehyde 16, a Wittig reaction with carbomethoxymethylenetriphenylphosphorane gave trans-alkene 17 and hydrolysis gave carboxylic acid 18.
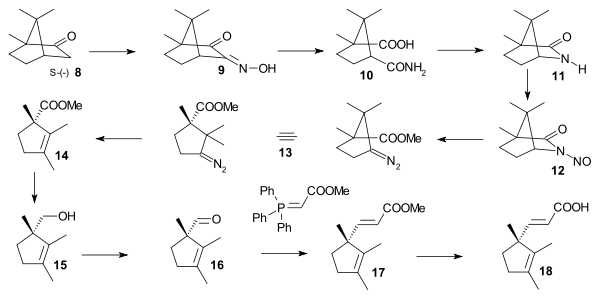 |
| Vitamin B12 D ring synthesis |
|---|
AD coupling
Amine 7 and carboxylic acid 18 were condensed through the acid chloride to amide 19. treatment with potassium tert-butoxide in tert-butanol then gave tricycle 20 in a Michael reaction with hydrogen atoms in trans relationship. In anticipation to the partial reduction of the aromatic ring in the later compound protective groups were added: one of the carbonyl groups as the ketal in 21 and the other as an enol ether through the iminium salt 22 (Triethyloxonium tetrafluoroborate) and the orthoamide 23 (sodium methoxide / methanol). Enol ether 24 was obtained by heating in toluene expelling ethanol. Birch reduction provided tetraene 25 and acid treatment gave the dione 26 dubbed pentacyclenone.
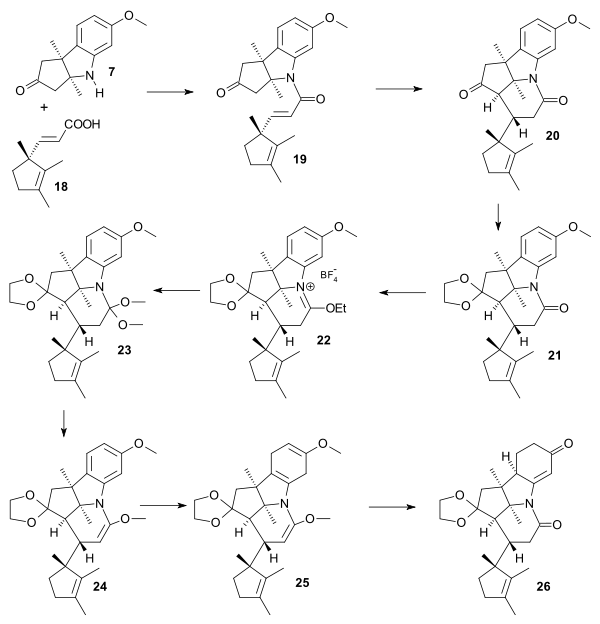 |
| Vitamin B12 AD ring synthesis |
|---|
The second protective group in 26 (acetal, acid hydrolysis) was reconverted to the ketone in 27. The monooxime 28 (at the more hindered ketone group) was synthesised from the dioxime by selective hydrolysis (nitrous acid / acetic acid ). The new nitrogen atom is also the second nitrogen atom required for the AD building block. Both the cyclopentene ring and the cyclohexenone ring were oxidized next in an ozonolysis (ozone) forming triketone 29, an aldol condensation of the 1,5-dicarbonyl unit (pyrrolidine acetate) formed cyclohexene 30 with tosylation of the oxime group, a second oxidation with periodic acid cleaves the cyclohexene ring and diazomethane esterifies the resulting carboxylic acid group in 31. A Beckmann rearrangement (methanol, sodium polystyrene sulfonate 2 hrs, 170 °C) took place next to lactam 32 (not isolated) which reacted further to the tetracycle 33 called alpha-corrnorsterone in an amine-carbonyl condensation - aldol condensation cascade. This compound resisted ring-opening of the lactam group due to incorrect stereochemistry of the propionic ester side-chain. The alpha compound was therefore converted its epimer 34 by first equilibrating in excess base followed by reacidifiying and diazomethane treatment. This epimer was then converted to the 35 by the simultaneous action of methanol and thiophenol. This ensured the differentiation of what will become the imidazole tail. The Ozonolysis gave aldehyde 36 with ammonia converting the thioester into an amide group and aldehyde reduction (sodium borohydride) followed by mesylation and bromination (lithium bromide) gave the bromide 37 with conversion of the amide group into a nitrile group as the completed AD section.
 |
| Vitamin B12 AD ring synthesis II |
|---|
Ring C synthesis
The starting material for the synthesis of ring C was chiral (+)-camphorquinone 38 [18] which can be converted to the acetoxy trimethylcyclohexene carboxylic acid ester 39 by addition of trifluoroborane in acetic anhydride, a reaction pioneered by Manasse & Samuel in 1902.[19] Ester hydrolysis to carboxylic acid 40 and amidation to amide 41 was followed by ozonolysis to peroxide 42 that was reduced to succinimide 43 by zinc and methanol. treatment with methanolic hydrochloric acid gave lactam 44 and pyrolysis gave the complete C ring 45.
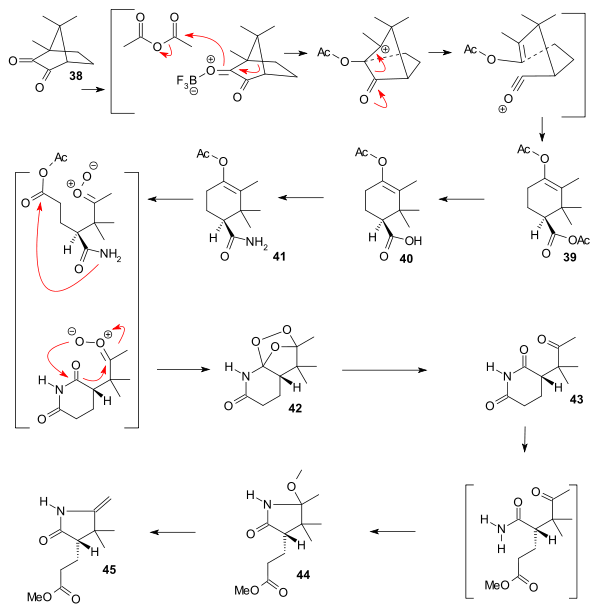 |
| Vitamin B12 C ring synthesis |
|---|
Ring B synthesis
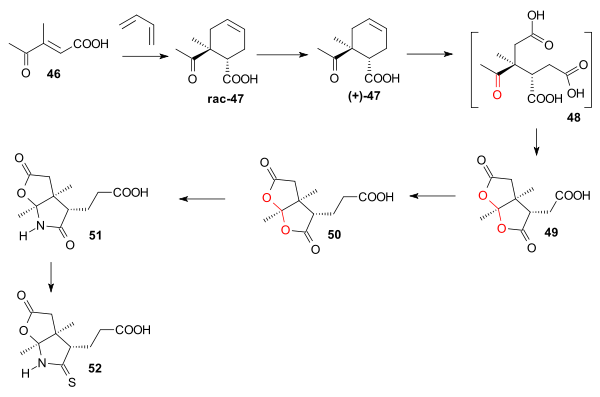
The starting material for ring B was 3-methyl-4-oxo-2-pentenoic acid 46 which was reacted with butadiene in a Diels-Alder reaction (stannic chloride) to racemic cyclohexene 47. This reaction is stereospecific with the methyl group and the carboxylic acid group ending up in a cis relationship. Chiral resolution using alpha-phenylethylamine gave optically active 47. Oxidation of the double bond with chromic acid gave the triacid 48 as an intermediate which gave dilactone 49 in two intramolecular esterifications. An Arndt–Eistert reaction elongated the carboxylic acid chain in 50, reaction with ammonia gave lactam 51 and reaction with phosphorus pentasulfide give thiolactam 52.
 |
| Vitamin B12 B ring synthesis |
|---|
BC coupling
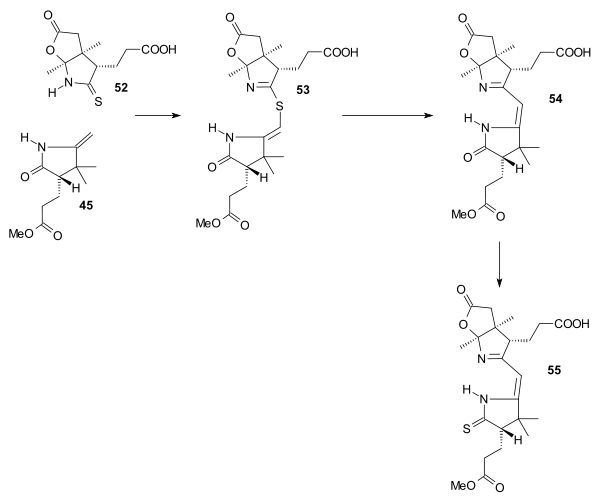
Ring B (52) and ring C (45) were joined with benzoyl peroxide/HCl to sulfur bridged 53, the sulfur atom was extruded with triethylphosphite to enamine-imine 54 in a first of two sulfur contractions and the lactam group converted to the thiolactam 55 (trimethyloxonium fluoroborate / hydrogen sulfide)
 |
| Vitamin B12 BC ring system synthesis |
|---|
AD BC coupling
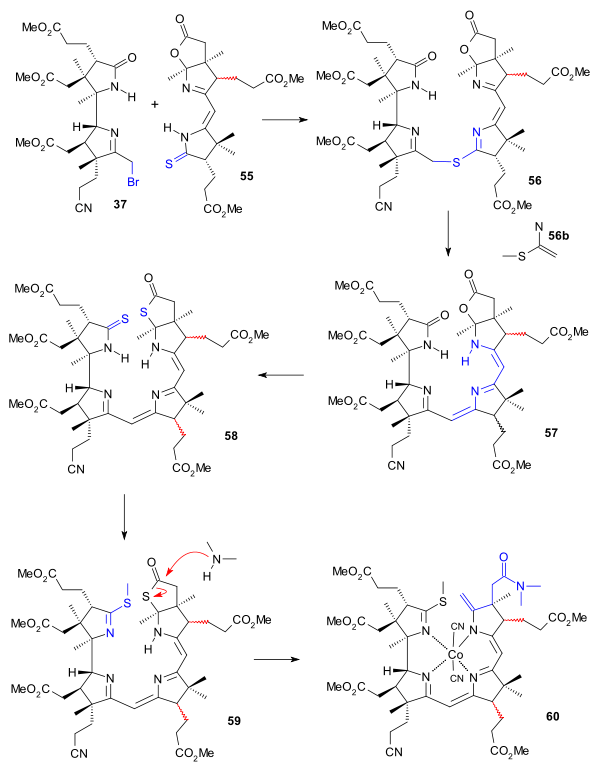
The eastern part of the molecule BC (cyanobromide 37) and the western part AD (thiodextrolin 55, with the propionic acid ester group racemized) were then joined using potassium t-butoxide to thioether 56 (through a sulfide ion intermediate). A second sulfur contraction (cyanoethyl phosphine/ trifluoroacetic acid/sulfolane) yielded cyanocorrigenolide 57 with the propionic acid ester group of ring C also racemised. Due to the steric bulk of both reactants this contraction was the only successful method. The oxygen atoms in the lactam and the lactone group were replaced by sulfur (phosphorus pentasulfide/4-methylpyridine) in dithiocyanocorrigenolide 58 and the S-methyl derivate 59 was formed by reaction with trimethyloxonium fluoroborate. Dimethylamine addition opened the thiolactone ring with an exocyclic alkene group in 60 by elimination of the sulfide anion from the methyl group. In an early example of template-directed synthesis this compound was isolated as the cobalt adduct.
 |
| Vitamin B12 BCAD construction |
|---|
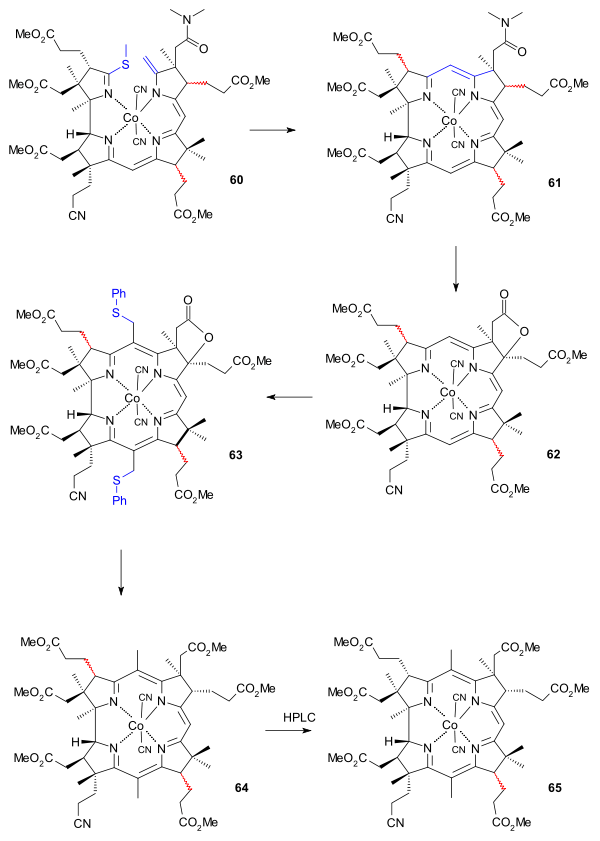
The final cyclisation reaction of 60 to 61 was facilitated by the central cobalt ion (placing the ends in close proximity) and consisted of another type of sulfur contraction employing basic conditions (diazabicyclononane / dimethylacetamide). This reaction takes place with racemisation of the propionic acid ester group of ring C. Oxidation (iodine/acetic acid) formed lactone 62 and restored the correct stereochemistry at the ring B propionic acid ester tail.
The final efforts were directed at placing methyl groups at position 5 and 15. With position 10 sufficiently shielded reaction with chloromethyl benzyl ether produced the di(chloromethyl) adduct which was further converted to the dithiophenyl compound 63 using thiophenol, isolation of which required thin layer chromatography. Desulfurisation took place with Raney nickel and the reduction reaction also opened the lactone ring to the carboxylic acid which was converted to the ester 64 by reaction with diazomethane. At this stage the number of isomers in the mixture was reduced by HPLC to just two with the racemic propionic acid ester group at C13 (ring C) remaining in 65. Reaction with sulfuric acid converted the cyano group to the amide group in 66, again destroying stereochemistry at C13. The correct isomer 67 (minor yield) was isolated again by HPLC.
 |
| Vitamin B12 BCAD construction part II |
|---|
The amide group was converted to the carboxylic acid group in 68 by action of the cyclohexylnitrone derived from chloroacetaldehyde in combination with silver tetrafluoroborate and in the final step 6 ester groups were converted to the amide groups in cobyric acid 69 by reaction with ammonia and ammonium chloride.
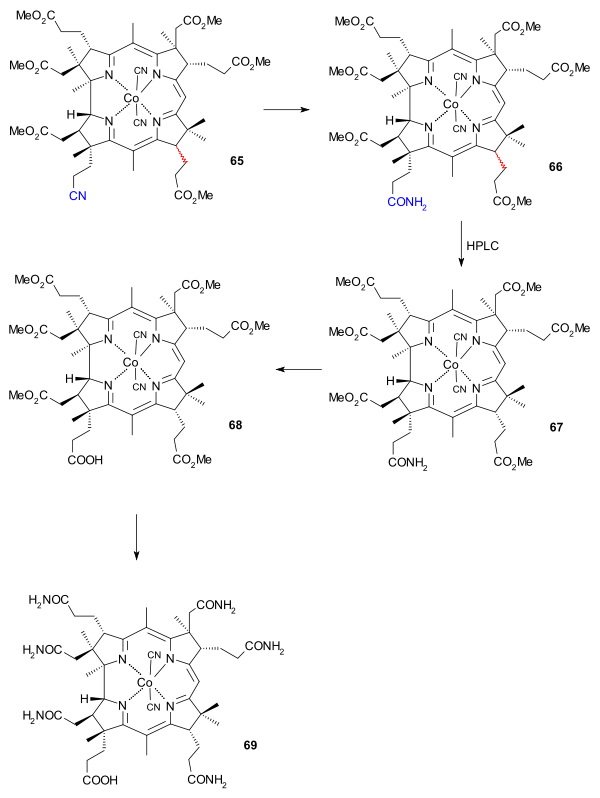 |
| Vitamin B12 BCAD construction part III |
|---|
External links
References
- 1 2 3 Woodward, R. B. (1968). "Recent advances in the chemistry of natural products". Pure Appl. Chem. 17: 519–547. doi:10.1351/pac196817030519.
- 1 2 3 Woodward, R. B. (1971). "Recent advances in the chemistry of natural products". Pure Appl. Chem. 25: 283–304. doi:10.1351/pac197125010283.
- 1 2 3 4 Woodward, R. B. (1973). "The total synthesis of vitamin B12". Pure Appl. Chem. 33 (1): 145–178. doi:10.1351/pac197333010145. PMID 4684454.
- 1 2 Eschenmoser, A. (1970). "Roads to Corrins". Quart. Rev. 24: 366–415. doi:10.1039/qr9702400366.
- 1 2 Eschenmoser, A. (1971). Studies on Organic Synthesis, in: XXIIIrd International Congress of Pure and Applied Chemistry: special lectures presented at Boston, USA, 26-30 July 1971, Vol. 2. London: Butterworths. pp. 69–106. doi:10.3929/ethz-a-010165162. ISBN 0-408-70316-4.
- 1 2 Eschenmoser, A. (1974). "Organische Naturstoffsynthese heute: Vitamin B12 als Beispiel". Naturwissenschaften. 61: 513–525. Bibcode:1974NW.....61..513E. doi:10.1007/BF00606511.
- 1 2 Eschenmoser, A.; Wintner, C. E. (1977). "Natural Product Synthesis and Vitamin B12". Science. 196 (4297): 1410–1420. doi:10.1126/science.867037. PMID 867037.
- 1 2 Nicolaou, K. C.; Sorensen, E. J. (1996). Classics in Total Synthesis: Targets, Strategies, Methods. Weinheim: VCH Verlag Chemie. ISBN 978-3-527-29231-8.
- ↑ Nicolaou, K. C.; Sorensen, E. J.; Winssinger, N. (1998). "The Art and Science of Organic and Natural Products Synthesis". J. Chem. Educ. 75: 1226–1258. doi:10.1021/ed075p1225.
- ↑ Nicolaou, K. C.; Vourloumis, D.; Winssinger, N.; Baran, P. S. (2000). "The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century". Angew. Chem. Int. Ed. 39 (1): 44–122. doi:10.1002/(SICI)1521-3773(20000103)39:1<44::AID-ANIE44>3.0.CO;2-L. PMID 10649349.
- ↑ Eschenmoser, A. (1987). "Vitamin B12: Experiments Concerning the Origin of Its Molecular Structure". Angew. Chem. Int. Ed. Engl. 27: 5–39. doi:10.1002/anie.198800051.
- ↑ Stevens, R. V. (1982). The Total Synthesis of Vitamin B12, in: Vitamin B12, Vol. 1 (D. Dolphin, ed.). New York: John Wiley & Sons. pp. 169–200. ISBN 978-0-471-03655-5.
- ↑ Jackson, A. H.; Smith, K. M. (1973). "The Total Synthesis of Pyrrole Pigments". Total Synth. Nat. Prod. 1: 143–278. doi:10.1002/9780470129647.ch3. ISBN 9780471032519.
- ↑ Mulzer, J.; Riether, D. (2003). "Total Synthesis of Cobyric Acid: Historical Development and Recent Synthetic Innovations". Eur. J. Org. Chem.: 30–45. doi:10.1002/1099-0690(200301)2003:1<30::AID-EJOC30>3.0.CO;2-I.
- ↑ Hodgkin, D. Crowfoot; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K.N.; White, J. G. (1956). "Structure of Vitamin B12". Nature. 178 (4524): 64–66. doi:10.1038/178064a0. PMID 13348621.
- ↑ Woodward, R. B. (1967). Aromaticity (Chemical Society Special Publication No. 21). London: Royal Society of Chemistry. pp. 217–249.
- ↑ Synthesen auf dem Vitamin-B12-Gebiet. 4. Mitteilung Partialsynthese von Vitamin B12 (p 704-712) W. Friedrich, G. Gross, K. Bernhauer, P. Zeller Helvetica Chimica Acta 1960 Volume 43 Issue 3, Pages 704 - 712 doi:10.1002/hlca.19600430314
- ↑ Camphorquinone: can be made from camphor by reaction with selenium dioxide see Organic Syntheses Coll. Vol. 10, p.204 (2004); Vol. 79, p.125 (2002). PDF
- ↑ Manasse, O.; Samuel, E. "Mittheilungen Reactionen des Campherchinons". Berichte der deutschen chemischen Gesellschaft. 35 (3): 3829–3843. doi:10.1002/cber.190203503216.