Atypical teratoid rhabdoid tumor
| Atypical teratoid rhabdoid tumor | |
|---|---|
|
MRI of an AT/RT | |
| Classification and external resources | |
| Specialty | oncology |
| ICD-10 | C70-C72 |
| ICD-9-CM | 191-192 |
| ICD-O | 9508/3 |
| OMIM | 609322 |
| DiseasesDB | 30780 |
| MedlinePlus | 000768 |
| eMedicine | ped/3012 |
| MeSH | D016543 |
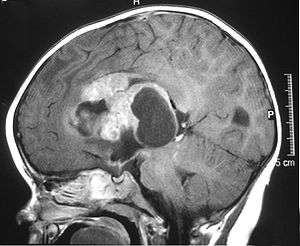
Atypical teratoid rhabdoid tumor (AT/RT) is a rare tumor usually diagnosed in childhood. Although usually a brain tumor, AT/RT can occur anywhere in the central nervous system (CNS) including the spinal cord. About 60% will be in the posterior cranial fossa (particularly the cerebellum). One review estimated 52% posterior fossa, 39% sPNET (supratentorial primitive neuroectodermal tumors), 5% pineal, 2% spinal, and 2% multi-focal.[1]
In the United States, three children per 1,000,000 or around 30 new AT/RT cases are diagnosed each year. AT/RT represents around 3% of pediatric cancers of the CNS.[2] Around 17% of all pediatric cancers involve the CNS; it is the most common childhood solid tumor. The survival rate for CNS tumors is around 60%. Pediatric brain cancer is the second leading cause of childhood death, just after leukemia. Recent trends suggest that the rate of overall CNS tumor diagnosis is increasing by about 2.7% per year. As diagnostic techniques using genetic markers improve and are used more often, the proportion of AT/RT diagnoses is expected to increase.
AT/RT was only recognized as an entity in 1996 and added to the World Health Organization (WHO) Brain Tumor Classification in 2000 (Grade IV).[3] The relatively recent classification and rarity has contributed to initial misdiagnosis and non-optimal therapy. This has led to a historically poor prognosis.[4]
Current research is focusing on using chemotherapy protocols that are effective against rhabdomyosarcoma in combination with surgery and radiation therapy.
Recent studies using multi-modal therapy have shown significantly improved survival data. In 2008, The Dana-Farber Cancer Institute in Boston reported two-year overall survival of 53% and event-free survival of 70% (median age at diagnosis of 26 months).[5] In 2013, The Medical University of Vienna reported five-year overall survival of 100%, and event-free survival of 89% (median age at diagnosis of 24 months).[6]
Survival rates can be significantly improved when the correct genetic diagnosis is made at the outset, followed with specific multi-modal treatment.
Classification
AT/RT may be related to malignant rhabdoid tumor (MRT), which occurs outside the CNS, usually in the kidney. The finding that AT/RT and MRT both have deletions of the INI1 gene indicates that rhabdoid tumors of the kidney and brain are at least closely related. AT/RT and MRT also have similar histology and similar clinical and demographic features. Moreover, 10–15% of MRT patients have simultaneous or subsequent brain tumors, many of which are secondary or primary MRT.
Signs and symptoms
Clinical signs and symptoms depend on the location of the tumor.
Since many of the tumors occur in the posterior fossa they present like other posterior fossa tumors, often with headache, vomiting, lethargy, and ataxia (unsteady gait). There is a case report of a seven-month-old child with a primarily spinal tumor that presented with progressive paraplegia and abnormal feeling in the legs.[7]
Genetics
Genetic similarities have been found within rhabdoid tumors. In particular the chromosomal 22 deletion is very common in AT/RTs. The chromosome 22 area contains the hSNF5/INI1 gene that appears to function as a classic tumor suppressor gene.[8] Most rhabdoid tumors have INI1 deletions whether they occur in the CNS, kidney or elsewhere. This mutation is viewed as the "first hit" which predisposes children to malignancies. INI1/hSNF5, a component of the chromatin remodeling SWI/SNF complex, is a critical tumor suppressor biallelically inactivated in rhabdoid tumors. Identification of INI1 as a tumor suppressor has facilitated accurate diagnosis of rhabdoid tumors.
The rate of transcription for SWI/SNF and HDAC complexes seem to be regulated by the INI1 gene. The SWI/SNF complex plays a role in chromatin remodeling. AT/RT is the first pediatric brain tumor for which a candidate tumor suppressor gene has been identified. A mutation or deletion in the INI1/hSNF5 gene occurs in the majority of AT/RT tumors. Up to 90% of AT/RT cases involve chromosome 22 deletion. This is mainly point mutations on the hSNF5/INI1 gene (i.e., one can diagnosis AT/RT without a chromosome 22 deletion elsewhere). The hSNF5/INI1 gene regulates 15 or so proteins in the chromatin structure. In addition, the OPN gene has a higher expression in AT/RT tumors. It is increasingly believed that the reason that all of the AT/RT cancers are not associated with the hSNF5/INI1 gene is that there are 14 additional proteins in the chromatin structure that are controlled by other genes. There are also some emerging mouse models of the AT/RT cancer as well as experimental cell lines derived from tumors. Despite these advances, the function of the gene is not yet understood. There is not enough known about the function of INI1, either as an independent modulator of gene expression or through its association with the SWI/SNF complex, to be able to use specific targeted biological agents for treatment. Prospective clinical and biologic trials are greatly needed to understand the efficacy of therapeutic interventions, as well as the role of the gene.
Risk for siblings and other members of the family
Atypical teratoid/rhabdoid tumors are very rare tumors and absolute risk to siblings is not reported in the literature. However, there have been some reports of AT/RTs presenting in two members of the same family, or one family member with an AT/RT and another with a renal rhabdoid tumor or other CNS tumor. These are suspected to arise from germ-line genetic mutations in a parent shared by affected siblings.
- A three-generation family is known in which two half-brothers were diagnosed with central nervous system atypical teratoid/rhabdoid tumors (AT/RT). The two boys, diagnosed at 2 months and 17 months of age, had a germline insertion mutation in exon 4 of the INI1 gene that was inherited from their healthy mother. A maternal uncle died in childhood from a brain tumor and a malignant rhabdoid tumor of the kidney. The identification of two unaffected carriers in a family segregating a germline mutation and rhabdoid tumor supports the hypothesis that there may be variable risks of development of rhabdoid tumor in the context of a germline mutation. There may be a developmental window in which most rhabdoid tumors occur. This family highlights the importance of mutation analysis in all patients with a suspected rhabdoid tumor.[9]
- In the first case report of monozygotic twins, both with brain tumors having similar genetic alterations, authors suggest a common genetic pathway.[10]
- A case reported on an infant that developed both AT/RT and renal rhabdoid tumors that were identical in gross and immunologic histology.[11]
- A family has had multiple generations of posterior fossa tumors including rhabdoid tumors and choroid plexus carcinoma. A germ-line mutation (SMARCB1) was found in both affected and some unaffected family members.[12]
- Two sisters were diagnosed with AT/RTs fifteen days apart. A case report stated there were no karyotypic anomalies noted.[13]
- Three siblings had a mutation of the SMARCB1 gene and one had a choroid plexus carcinoma and two had an AT/RT. Although the mother had a normal somatic DNA it appears that the mutation was inherited from the mother's germline due to a mutation during oogenesis.[14]
- Izycka-Swieszewska et al. describe a five-month-old child with an AT/RT, whose father was diagnosed with a primitive neuroectodermal tumor (PNET) of the spinal canal. FISH analysis showed significant genetic differences in the specimens which suggest that the occurrence of these virulent CNS malignancies within a single family was coincidental.[15]
Pathology
Pathology is the study and diagnosis of disease through examination of organs, tissues, bodily fluids and whole bodies (Autopsy). AT/RT and rhabdoid tumor share the term "rhabdoid" because under a microscope both tumors resemble rhabdomyosarcoma.
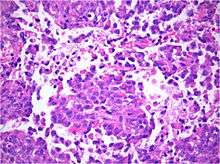
Histology
Histology is the study of the microscopic anatomy of cells and tissues of plants and animals. It is performed by examining a thin slice of tissue under a light microscope. The tumor histopathology is jumbled small and large cells. The tissue of this tumor contains many different types of cells including the rhabdoid cells, large spindled cell, epithelial and mesenchymal cells and areas resembling primitive neuroectodermal tumor (PNET). As much as 70% of the tumor may be made up of PNET-like cells. Ultrastructure characteristic whorls of intermediate filaments in the rhabdoid tumors (as with rhabdoid tumors in any area of the body). Ho and associates found sickle-shaped embracing cells, previously unreported, in all of 11 cases of AT/RT.[16]
Immunohistochemistry
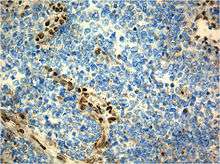
Immunohistochemistry refers to the process of localizing proteins in cells of a tissue section exploiting the principle of antibodies binding specifically to antigens in biological tissues. A tissue sample is stained to identify specific cellular proteins. Immunohistochemical staining is widely used in the diagnosis and treatment of cancer. Specific molecular markers are characteristic of particular cancer types. Immunohistochemistry is also widely used in basic research to understand the distribution and localization of biomarkers in different parts of a tissue. Proteins found in an Atypical Teratoid Rhaboid Tumor are:
- Vimentin-positive
- Cytokeratin-positive
- Neuron specific enolase-positive
- Epitelial membrane antigen-positive
- Glial fibrillary acidic protein- positive
- Synaptophysin
- Chromogranin
- Smooth muscle actin
- Desmin
- Carcinoembrionary antigen
- CD99 antigen;[17][18]
- S-100
- neurofilaments
- AFP – not found
- HCG – negative
Cytogenetic studies
Cytogenetics is the study of a tumor’s genetic make-up. A technique called FISH may be able to help locate a mutation or abnormality that may be allowing tumor growth.[19] This technique has been shown to be useful in identifying some tumors and distinguishing two histologically similar tumors from each other (such as AT/RTs and PNETs). In particular, medulloblastmas/PNETs may possibly be differentiated cytogenetically from AT/RTs as chromosomal deletions of 17p are relatively common with medulloblastoma and abnormalities of 22q11.2 are not seen. On the other hand, chromosomal 22 deletions are very comomon in AT/RTs.
In importance of the hSNF5/INI1 gene located on chromosomal band 22q11.2 is highlighted in the summary paper form the Workshop on Childhood Atypical Teratoid Rhabdoid Tumors as the mutation’s presence is sufficient to change the diagnosis from a medulloblastoma or PNET to the more aggressive AT/RT classification. However, it should be noted that this mutation is not present in 100% of cases. Therefore, if the mutation is not present in an otherwise classic AT/RT immunohistochemical and morphologic pattern then the diagnosis remains an AT/RT.
Diagnosis
The standard work-up for AT/RT includes:
- Magnetic resonance imaging (MRI) of the brain and spine
- Lumbar puncture to look for M1 disease
- Computed tomography (CT) of chest and abdomen to check for a tumor
- Bone marrow aspiration to check for bone tumors. Sometimes the physician will perform a stem cell transplant
- Bone marrow biopsy
- Bone scan
The initial diagnosis of a tumor is made with a radiographic study (MRI[20] or CT-). If CT was performed first, an MRI is usually performed as the images are often more detailed and may reveal previously undetected metastatic tumors in other locations of the brain. In addition, an MRI of the spine is usually performed. The AT/RT tumor often spreads to the spine. It is difficult to diagnose AT/RT only from radiographic study; usually a pathologist must perform a cytological or genetic analysis.
Examination of the cerebrospinal fluid (CSF) is important as one-third of patients will have intracranial dissemination with involvement of the CSF. Large tumor cells, eccentricity of the nuclei and prominent nucleoli are consistent findings.[21] Usually only a minority of AT/RT biopsies have rhabdoid cells, making diagnosis more difficult. Increasingly it is recommended that a genetic analysis be performed on the brain tumor, especially to find if a deletion in the INI1/hSNF5 gene is involved (appears to account for over 80% of the cases). The correct diagnosis of the tumor is critical to any protocol. Studies have shown that 8% to over 50% of AT/RT tumors are diagnosed incorrectly.
Differential diagnosis
The critical step in treatment planning is to determine the correct histology of the tumor. Misidentification of the tumor histology can lead to errors in treatment and prognosis.[22]
Atypical teratoid/rhaboid tumor closely resembles medulloblastoma,[23] primitive neuroectodermal tumor, choroid plexus carcinoma, and some kinds of germ cell tumor. Because rhabdoid characteristics are not the only component of AT/RT, some sections of an AT/RT may resemble other tumors. These characteristics may be present only in focal areas or may be less pronounced.
It is important to consider AT/RT when a medulloblastoma or PNET is suspected, particularly in a child under the age of one. Cytogenetic studies can assist in differentiating MB/PNETs from AT/RTs. Some kinds of germ cell tumor secrete tumor markers AFP or bHCG; AT/RTs do not.
Compared to medulloblastoma, AT/RT has a significantly worse prognosis. AT/RT occurs in young children (often younger than three years) who are difficult to evaluate, it is resistant to many current therapies, and its recurrence is fast.
Appearance on radiologic exam
AT/RTs can occur at any sites within the CNS; however, approximately 60% are located in the posterior fossa or cerebellar area. The ASCO study showed 52% posterior fossa; 39% sPNET (supratentorial primitive neuroectodermal tumors); 5% pineal; 2% spinal, and 2% multi-focal.[1]
The tumors' appearance on CT and MRI are nonspecific, tending towards large size, calcifications, necrosis (tissue death),and hemorrhage (bleeding). Radiological studies alone cannot identify AT/RT; a pathologist almost always has to evaluate a brain tissue sample.
The increased cellularity of the tumor may make the appearance on an uncontrasted CT to have increased attenuation. Solid parts of the tumor often enhance with contrast MRI finding on T1 and T2 weighted images are variable. Pre-contrast T2 weighted images may show an iso-signal or slightly hyper-signal. Solid components of the tumor may enhance with contrast but do not always. MRI studies appear to be more able to pick up metastatic foci in other intracranial locations as well as intraspinal locations.
Preoperative and follow-up studies are needed to detect metastatic disease.
Treatment
Surgery
Surgery plays a critical role in obtaining tissue to make an accurate diagnosis. Surgery alone is not curative. In addition, 30% of the AT/RTs are located supratentorially and there is a predilection for the cerebello-pontine angle[24] which makes surgical resection difficult. One-third or more children will have disseminated disease at the time of diagnosis. Total or near-total resections are often not possible.
Chemotherapy
Approximately 50% of the AT/RTs will transiently respond, but chemotherapy by itself is rarely curative. There is no standard treatment for AT/RT. Various chemotherapeutic agents have been used against AT/RTs which are also used against other CNS tumors including cisplatinum, carboplatinum, cyclophosphamide, vincristine and etoposide. Some Chemotherapy protocols are listed below:
- CCG clinical trial CCG-9921 was activated in 1993 and published its results in 2005. The proposed treatments did not have different outcomes and were not an improvement on prior treatments.[25] Geyer published a review of chemotherapy on 299 infants with CNS tumors that evaluated response rate, event-free survival (EFS), and toxicity of two chemotherapeutic regimens for treatment of children younger than 36 months with malignant brain tumors. Patients were randomly assigned to one of two regimens of induction chemotherapy (vincristine, cisplatin, cyclophosphamide, and etoposide v vincristine, carboplatin, ifosfamide, and etoposide). Intensified induction chemotherapy resulted in a high response rate of malignant brain tumors in infants. Survival was comparable to that of previous studies, and most patients who survived did not receive radiation therapy.[25]
- Sarcoma protocols. There has been at least one report in the literature of malignant rhabdoid tumors of the CNS being treated in as a high-grade intracranial sarcoma. These three cases were treated with surgery, chemotherapy, radiotherapy and triple intrathecal chemotherapy similar to the Intergroup Rhabdomyosarcoma Study III guidelines.[26]
- Intrathecal protocols. One of the difficulties with brain and spinal tumors is that the blood brain barrier needs to be crossed so that the drug can get to the tumor. One mechanism to deliver the drug is through a device called an Ommaya reservoir. This is a device which shares some characteristics with a shunt in which a tube a surgically placed in the fluid surrounding the brain and a bulb shaped reservoir attached to the tubing is placed under the skin of the scalp. When the child is to receive intrathecal chemotherapy, the drug is administered into this bulb reservoir. At other times intrathecal chemotherapeutic agents are delivered through a lumbar puncture (spinal tap). A current Pediatric Brain Tumor Consortium Protocol uses intrathecal mafosfamide, a pre-activated cyclophosphamide derivative, in addition to other modalities to try to effect this tumor.[27]
- High dose chemotherapy with stem cell rescue. This therapy uses chemotherapy at doses high enough to completely suppress the bone marrow. Prior to instituting this therapy, the child has a central line placed and stem cells are gathered. After therapy these cells are given back to the child to regrow the bone marrow. Stem cell rescue or autologous bone marrow transplantation, was initially thought to be of benefit to a wide group of patients, but has declined over the history of chemotherapy protocols.
Radiation therapy
The traditional practice for childhood brain tumors has been to use chemotherapy and to defer radiation therapy until a child is older than three years. This strategy is based upon observations that children under three have significant long term complications as a result of brain irradiation. However, the long term outcomes of AT/RT are so poor that some protocols call for upfront radiation therapy, often in spite of young age.[28]
The dose and volume of radiation had not been standardized, however, radiation does appear to improve survival. The use of radiation has been limited in children younger than three because of the risk of severe neurocognitive deficits. There are protocols using conformal, local radiation in the young child to try to cure this tumor.
External beam (conformal) radiation uses several beams that intersect at the tumor location; the normal brain tissue receives less radiation and cognitive function is thereby less affected.
Proton beam radiation was only offered at Massachusetts General Hospital in Boston and at Loma Linda, California as of 2002. Since 2003 three or four more proton therapy centers have opened in the United States. St. Jude Children's Research Hospital is in the process of building one at their Memphis, Tn. location. Some centres have since opened in Europe. (Germany, Switzerland and France).[29][30][31][32][33][34]
Chromatin re-modeling agents
This protocol is still in pre-clinical evaluation. HDAC inhibitors are a new class of anticancer agents targeted directly at chromatin remodeling. These agents have been used in acute promyelocytic leukemia and have been found to affect the HDAC-mediated transcriptional repression. There is too little understanding of the INI1 deficiency to predict whether HDAC inhibitors will be effective against AT/RTs. There are some laboratory results that indicate it is effective against certain AT/RT cell lines.[35]
Prognosis
The prognosis for AT/RT has been very poor, although there are some indications that an IRSIII-based therapy can produce long-term survival (60 to 72 months). Two-year survival is less than 20%, average survival postoperatively is 11 months, and doctors often recommend palliative care, especially with younger children because of the poor outcomes. Recently a protocol utilized by a multicenter trial reported in the Journal of Clinical Oncology resulted in a 70% survival rate at 2–3 years, with most relapses occurring within months, leading to hope that there is a point beyond which patients can be considered cured.[36]
Patients with metastasis (disseminated tumor), larger tumors, tumors that could not be fully removed, or tumor recurrence, and who were younger than 36 months had the worst outcomes (i.e., shorter survival times).
A retrospective survey from 36 AT/RT St. Jude Children's Hospital patients from 1984 to 2003 showed that the Two-year event-free survival (EFS) for children under three was 11%, the overall survival (OS) rate was 17%. For children aged 3 years or older the EFS was 78% and the OS 89%.[4] A retrospective register at the Cleveland Children's hospital on 42 AT/RT patients found median survival time is 16.25 months and a survival rate around 33%. One-quarter of these cases did not show the mutation in the INI1/hSNF5 gene.
The longest term survivals reported in the literature are:
- (a) Hilden and associates reported a child who was still free from disease at 46 months from diagnosis.[37]
- (b) Olson and associates reported a child who was disease free at five years from diagnosis based on the IRS III protocol.[38]
- (c) In 2003 Hirth reported a case who had been disease free for over six years.[39]
- (d) Zimmerman in 2005 reported 50-to-72 month survival rates on four patients using an IRS III-based protocol. Two of these long term survivors had been treated after an AT/RT recurrence.[40]
- (e) A NYU study (Gardner 2004) has 4 of 12 longer term AT/RT survivors; the oldest was alive at 46 months after diagnosis.[41]
- (f) Aurélie Fabre, 2004, reported a 16-year survivor of a soft-tissue rhabdoid tumor.[42]
- (g) Medical University of Vienna, 2013, reported a 16-year survivor, among other long-term survivors [6]
Cancer treatments in long-term survivors who are children usually cause a series of negative effects on physical well being, fertility, cognition, and learning.[43][44][45][46]
Metastasis
Metastatic spread is noted in approximately one-third of the AT/RT cases at the time of diagnosis and tumors can occur anywhere throughout the CNS. The ASCO study of the 188 documented AT/RT cases prior to 2004 found 30% of the cases had metastasis at diagnosis.[1] Metastatic spread to the meninges (leptomenigeal spread sometimes referred to as sugar coating) is common both initially and with relapse. Average survival times decline with the presence of metastasis. Primary CNS tumors generally metastasize only within the CNS.
One case of metastatic disease to the abdomen via ventriculoperitoneal shunt has been reported with AT/RT . Metastatic dissemination via this mechanism has been reported with other brain tumors including germinomas, medulloblastomas, astrocytomas, glioblastomas, ependymomas and endodermal sinus tumors. Guler and Sugita separately reported cases of lung metastasis without a shunt.[47][48]
Epidemiology
An estimated 3% of pediatric brain tumors are AT/RTs although this percentage may increase with better differentiation between PNET/medulloblastoma tumors and AT/RTs.
As with other CNS tumors, slightly more males are affected than females (ratio 1.6:1). The ASCO study showed a 1.4:1 male to female ratio.[1]
History
Atypical teratoid/rhabdoid tumor was first described as a distinct entity in 1987.[49] Before 1978, when rhabdoid tumor was described, AT/RT likely was misdiagnosed as medulloblastoma. In some early reports the tumor was known also as malignant rhabdoid tumor (MRT) of the CNS. Between 1978 and 1987, AT/RT was usually misdiagnosed as rhabdoid tumor. However, both AT/RT and non-CNS MRT have a worse prognosis than medulloblastoma and are resistant to the standard treatment protocols for medulloblastoma.
By 1995, AT/RT had become regarded as a newly defined aggressive, biologically unique class of primarily brain and spinal tumors, usually affecting infants and young children.[50] In January 2001, the U.S. National Cancer Institute and Office of Rare Diseases hosted a Workshop on Childhood Atypical Teratoid/Rhabdoid Tumors of the Central Nervous System. Twenty-two participants from 14 institutions came together to discuss the biology, treatments and new strategies for these tumors. The consensus paper on the biology of the tumor was published in Clinical Research.[51] The workshop's recognition that CNS atypical teratoid/rhabdoid tumors (AT/RT) have deletions of the INI1 gene indicates that rhabdoid tumors of the kidney and brain are identical or closely related entities. This observation is not surprising because rhabdoid tumors at both locations possess similar histologic, clinical, and demographic features.
Research directions
Atypical teratoid rhabdoid tumor is rare and no therapy has been proven to deliver long-term survival, nor is there a set of standard protocols. Thus, most children with AT/RT are enrolled in clinical trials to attempt to find an effective cure. A clinical trial is not a treatment standard; it is research. Some clinical trials compare an experimental treatment to a standard treatment, but only if such a standard treatment exists.
There is an ongoing research into stem cell transplant surgeries.
Cultural references
In 2011, The New Yorker published an article by Aleksandar Hemon, about the author's daughter's battle with AT/RT.[52]
In August 2011, a 6-year-old named Avalanna Routh who was battling AT/RT at Dana–Farber Cancer Institute was given a pretend wedding with her idol Justin Bieber, with doctors and nurses providing a cardboard life-sized cutout of Bieber, a guitarist, flowers, and a T-shirt emblazoned with the words "Future Mrs. Bieber". In February 2012, she spent the day in person with Justin Bieber, her pretend husband, after a Facebook campaign to meet her idol.[53] On September 26, 2012, she died after battling AT/RT for five and a half years.[54]
The video game That Dragon, Cancer is based on the experiences of Ryan and Amy Green raising their son Joel after he was diagnosed with an atypical teratoid rhabdoid tumor at 12 months and given only about four months to live. Joel continued to survive for four more years after suffering from seven additional tumors and eventually succumbing to cancer on March 13, 2014. Ryan Green wanted to provide the experiences of raising Joel in the form of a video game to help the player to understand the difficulties and realities they had to deal with during this time.[55][56] After Joel's death, the game was reworked to instead act as a tribute to Ryan and Amy's five short years with their third child.
See also
References
- 1 2 3 4 Kieran MW (2006). "An Update on Germ Cell Tumors, Atypical Teratoid/Rhaboid Tumors, and Choroid Plexus Tumors Rare Tumors 3: Brain Tumors---Germ Cell Tumors, Atypical Teratoid/Rhabdoid Tumors, and Choroid Plexus Tumors". American Society of Clinical Oncology. Education. Book. Archived from the original on 2008-01-07. Retrieved 2007-05-20.
- ↑ Measure D6: Types of Childhood Cancer – 2006 Tables D6a & D6b. U.S. Environmental Protection Agency. Retrieved on 2008-04-17.
- ↑ Kleihues, P. (2000). Pathology and genetics of tumours of the nervous system. IARC Press: Lyon 2000. ISBN 92 83 22409 4.
- 1 2 Tekautz TM, Fuller CE, Blaney S, et al. (2005). "Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy". J. Clin. Oncol. 23 (7): 1491–9. doi:10.1200/JCO.2005.05.187. PMID 15735125. See Figure 1.
- ↑ Chi, Susan (2008). "Intensive Multimodality Treatment for Children With Newly Diagnosed CNS Atypical Teratoid Rhabdoid Tumor". Journal of Clinical Oncology. 27: 385–389. doi:10.1200/JCO.2008.18.7724.
- 1 2 Slavc, Irene (2014). "Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012". Cancer Medicine. 3 (1): 91–100. doi:10.1002/cam4.161.
- ↑ Tamiya T, Nakashima H, Ono Y, et al. (2000). "Spinal atypical teratoid/rhabdoid tumor in an infant". Pediatr Neurosurg. 32 (3): 145–9. doi:10.1159/000028920. PMID 10867562. Archived from the original on 2011-05-17.
- ↑ Chung-Lan Kao; Shih-Hwa Chiou; Yann-Jang Chen; Sher Singh; Han-Tso Lin; Ren-Shyan Liu; Chih-Wen Lo; Chi-Chang Yang; Chin-Wen Chi; Chen-hsen Lee; Tai-Tong Wong (2005). "Increased expression of osteopontin gene in atypical teratoid/rhabdoid tumor of the central nervous system". Modern Pathology. 18 (6): 769–778. doi:10.1038/modpathol.3800270. PMID 15776015. Retrieved 2008-05-05.
- ↑ Janson K, Nedzi LA, David O, et al. (2006). "Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation". Pediatr Blood Cancer. 47 (3): 279–84. doi:10.1002/pbc.20622. PMID 16261613.
- ↑ Fernandez, C; Bouvier, C; Sévenet, N; Liprandi, A; Coze, C; Lena, G; Figarella-Branger, D (2002). "Congenital disseminated malignant rhabdoid tumor and cerebellar tumor mimicking medulloblastoma in monozygotic twins: pathologic and molecular diagnosis". Am J Surg Pathol. 26 (26:): 266–70. doi:10.1097/00000478-200202000-00016. PMID 11812951.
- ↑ Beigel JA; Fogelgren B; Wainwright LM; et al. (2000). "Germ-line INI1 mutations in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor". Genes Chromosomes Cancer. 28 (1): 31–7. doi:10.1002/(SICI)1098-2264(200005)28:1<31::AID-GCC4>3.0.CO;2-Y. PMID 10738300.
- ↑ Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000). "Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene". Am. J. Hum. Genet. 66 (4): 1403–6. doi:10.1086/302833. PMC 1288204
 . PMID 10739763.
. PMID 10739763. - ↑ Proust F, Laquerriere A, Constantin B, Ruchoux MM, Vannier JP, Fréger P (1999). "Simultaneous presentation of atypical teratoid/rhabdoid tumor in siblings" (PDF). J. Neurooncol. 43 (1): 63–70. doi:10.1023/A:1006114732613. PMID 10448873.
- ↑ Sevent N, Sheridan E, Amran D, et al. (1999). "Constitutional mutations of the hSNF/INI1 gene predispose to a variety of cancers". Am J Hum Genet (65): 1343–48.
- ↑ Ewa Izycka-Swieszewska; Maria Debiec-Rychter; Bartosz Wasag; et al. (February 2003), "A unique occurrence of a cerebral atypical teratoid/rhabdoid tumor in an infant and a spinal canal primitive neuroectodermal tumor in her father", Journal of Neuro-Oncology, 61 (3): 219–225, doi:10.1023/A:1022532727436
- ↑ Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H (2000). "Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma". Acta Neuropathol. 99 (5): 482–8. doi:10.1007/s004010051149. PMID 10805090.
- ↑ "CD99". Ncbi.nlm.nih.gov. 2013-01-30. Retrieved 2013-02-22.
- ↑ "2nd CD99 link". Ncbi.nlm.nih.gov. Retrieved 2013-02-22.
- ↑ Bruch LA, Hill DA, Cai DX, Levy BK, Dehner LP, Perry A (2001). "A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors". Hum. Pathol. 32 (2): 156–62. doi:10.1053/hupa.2001.21572. PMID 11230702.
- ↑ Meyers SP, Khademianc ZP, Biegeld JA, Chuange SH, Koronesb DN, Zimmerman RA (May 2006). "Primary Intracranial Atypical Teratoid/Rhabdoid Tumors of Infancy and Childhood: MRI Features and Patient Outcomes". American Journal of Neuroradiology. 27 (5): 962–971. PMID 16687525. Retrieved 2008-05-05.
- ↑ Lu L, Wilkinson EJ, Yachnis AT (2000). "CSF cytology of atypical teratoid/rhabdoid tumor of the brain in a two-year-old girl: a case report". Diagn. Cytopathol. 23 (5): 329–32. doi:10.1002/1097-0339(200011)23:5<329::AID-DC9>3.0.CO;2-W. PMID 11074628.
- ↑ Jay V, Edwards V, Halliday W, Rutka J, Lau R (1997). ""Polyphenotypic" tumors in the central nervous system: problems in nosology and classification". Pediatr Pathol Lab Med. 17 (3): 369–89. doi:10.1080/107710497174697. PMID 9185218.
- ↑ Burger PC, Yu IT, Tihan T, et al. (1998). "Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study". Am. J. Surg. Pathol. 22 (9): 1083–92. doi:10.1097/00000478-199809000-00007. PMID 9737241.
- ↑ "PDF" (PDF). Retrieved 2013-02-22.
- 1 2 Geyer JR, Sposto R, Jennings M, et al. (2005). "Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group". J. Clin. Oncol. 23 (30): 7621–31. doi:10.1200/JCO.2005.09.095. PMID 16234523.
- ↑ "Childhood Rhabdomyosarcoma Treatment". National Cancer Institute. Archived from the original on 4 July 2007. Retrieved 2007-07-09.
- ↑ Poussaint TY, Phillips PC, Vajapeyam S, et al. (2007). "The Neuroimaging Center of the Pediatric Brain Tumor Consortium-collaborative neuroimaging in pediatric brain tumor research: a work in progress". AJNR. American journal of neuroradiology. 28 (4): 603–7. PMID 17416804.
- ↑ Squire SE, Chan MD, Marcus KJ (2007). "Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy". J. Neurooncol. 81 (1): 97–111. doi:10.1007/s11060-006-9196-z. PMID 16855864.
- ↑ "Principles of Proton Beam Therapy". Massgeneral.org. Retrieved 2013-02-22.
- ↑ Christopher Owen - Pageservant. "Proton Beam Radiotherapy at Mass. General". Neurosurgery.mgh.harvard.edu. Retrieved 2013-02-22.
- ↑ Proton Beam Therapy Article Archived October 8, 2007, at the Wayback Machine.
- ↑ British Journal of Cancer (2009-06-01). "Proton Beam Therapy - BJC Abstract". Nature.com. Retrieved 2013-02-22.
- ↑ Loma Linda Medical Center Proton Treatment Center - Overview Archived February 6, 2007, at the Wayback Machine.
- ↑ Loma Linda overview of Childhood Brain Tumors Archived April 12, 2007, at the Wayback Machine.
- ↑ Zhang ZK, Davies KP, Allen J, et al. (2002). "Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5". Mol. Cell. Biol. 22 (16): 5975–88. doi:10.1128/MCB.22.16.5975-5988.2002. PMC 133966. PMID 12138206.
- ↑ "New hope for patients with rare brain tumors". Danafarberchildrens.org. 2009-03-10. Retrieved 2013-02-22.
- ↑ Hilden JM, Meerbaum S, Burger P, et al. (2004). "Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry". J. Clin. Oncol. 22 (14): 2877–84. doi:10.1200/JCO.2004.07.073. PMID 15254056. Retrieved 2007-05-23.
- ↑ Olson TA, Bayar E, Kosnic E (1995). "Successful treatment of disseminated central nervous system malignant rhabdoid tumors". J Pediatr Hematol Oncol. 17 (1): 71–75. doi:10.1097/00043426-199502000-00013. PMID 7743242.
- ↑ Hirth A, Pedersen PH, Wester K, et al. (2003). "Cerebral Atypical Teratoid/Rhabdoid Tumor of Infancy: Long-Term Survival after Multimodal Treatment, also Including Triple Intrathecal Chemotherapy and Gamma Knife Radiosurgery--Case Report (Abstract)". Pediatric Hematology and Oncology 2003. 20 (4): 327–332. doi:10.1080/713842315.
- ↑ Zimmerman MA, Goumnerova LC, Proctor M, et al. (2005). "Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor". J. Neurooncol. 72 (1): 77–84. doi:10.1007/s11060-004-3115-y. PMID 15803379. Retrieved 2007-05-20.
- ↑ Gardner S, Diez B, Green A, et al. (June 2004). "THER 27. INTENSIVE INDUCTION CHEMOTHERAPY FOLLOWED BY HIGH-DOSE CHEMOTHERAPY WITH AUTOLOGOUS STEM CELL RESCUE (ASCR) IN YOUNG CHILDREN NEWLY DIAGNOSED WITH CENTRAL NERVOUS SYSTEM (CNS) ATYPICAL TERATOID RHABDOID TUMORS (ATT/RT)—THE "HEAD START" REGIMENS" (PDF). Abstracts from the Eleventh International Symposium on Pediatric Neuro-Oncology. Retrieved 2007-06-03.
- ↑ Fabre A, Eyden B, Ali HH (January 2004). "Soft-Tissue Extrarenal Rhabdoid Tumor with a Unique Long-Term Survival". Ultrastructural Pathology. 28 (1): 49–52. doi:10.1080/01913120490275259. PMID 14967599. Retrieved 2007-05-28.
- ↑ Fouladi M, Gilger E, Kocak M, et al. (October 1, 2005). "Intellectual and Functional Outcome of Children 3 Years Old or Younger Who Have CNS Malignancies". Journal of Clinical Oncology. 23 (28): 7152–60. doi:10.1200/JCO.2005.01.214. PMID 16192599.
- ↑ Monteleone P, Meadows AT (June 6, 2006). "Late Effects of Childhood Cancer and Treatment". EMedicine from WebMD.
- ↑ Foreman NK, Faestel PM, Pearson J, et al. (January 1999). "Health Status in 52 Long-term Survivors of Pediatric Brain Tumors". Journal of Neuro-Oncology. 41 (1): 47–52. doi:10.1023/A:1006145724500.
- ↑ Meyers EA, Kieran MW (2002). "Brief Report Psychological adjustment of surgery-only pediatric neuro-oncology patients: a retrospective analysis". Psycho-Oncology. John Wiley & Sons, Ltd. 11 (1): 74–79. doi:10.1002/pon.553. PMID 11835594.
- ↑ Güler E, Varan A, Söylemezoglu F, et al. (2001). "Extraneural metastasis in a child with atypical teratoid rhabdoid tumor of the central nervous system" (PDF). J. Neurooncol. 54 (1): 53–6. doi:10.1023/A:1012540700093. PMID 11763423.
- ↑ Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M (1999). "Pineal malignant rhabdoid tumor with chondroid formation in an adult". Pathol. Int. 49 (12): 1114–8. doi:10.1046/j.1440-1827.1999.00988.x. PMID 10632935.
- ↑ Rorke LB, Packer RJ, Biegel JA (1996). "Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity". J. Neurosurg. 85 (1): 56–65. doi:10.3171/jns.1996.85.1.0056. PMID 8683283.
- ↑ Rorke LB, Packer R, Biegel J (1995). "Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood". J. Neurooncol. 24 (1): 21–8. doi:10.1007/BF01052653. PMID 8523069.
- ↑ Biegel JA, Kalpana G, Knudsen ES, et al. (2002). "The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors". Cancer Res. 62 (1): 323–8. PMID 11782395.
- ↑ Hemon, Aleksandar (June 13, 2011). "The Aquarium". The New Yorker. Retrieved March 2, 2012.
- ↑ "Justin Bieber Meets With Merrimac Girl Battling Brain Cancer". CBS Boston. CBS Local Media, a division of CBS Radio Inc. February 13, 2012. Retrieved March 2, 2012.
- ↑ "'Mrs. Bieber' Avalanna Routh dies at age 6". CNN.com. September 27, 2012. Retrieved October 26, 2012.
- ↑ Robertson, Andy (July 12, 2013). "That Dragon, Cancer: the video game helping a father face his son's disease". The Daily Telegraph. Retrieved March 13, 2014.
- ↑ Futter, Mike (March 13, 2014). "Joel Green, Inspiration For That Dragon, Cancer, Passes Away At Age 5". Game Informer. Retrieved March 13, 2014.
Further reading
- Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Walter AW, Rorke LB, Biegel JA (July 2004). "Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry". J. Clin. Oncol. 22 (14): 2877–84. doi:10.1200/JCO.2004.07.073. PMID 15254056.
- Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, Weissman B, Smith M (January 2002). "The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors". Cancer Res. 62 (1): 323–8. PMID 11782395.
- Huret J, Sevenet N (2002). "Rhabdoid predispostion syndrome". Atlas of Genetics and Cytogenetics in Oncology and Haematology. Archived from the original on 2005-12-26.
External links
- iSTAR AT/RT Initiative for the Study and Translational AT/RT Research
- CureATRT.org - A community for those fighting atypical teratoid rhabdoid tumors
- Rhabdoid Tumor - Frequently Asked Questions