Leukodystrophy
 | |
| T2 weighted axial scan at the level of the caudate heads demonstrates marked loss of posterior white matter, with reduced volume and increased signal intensity. The anterior white matter is spared. Features are consistent with X-linked adrenoleukodystrophy. | |
| Classification and external resources | |
|---|---|
| Specialty | endocrinology |
| ICD-10 | E75.2 |
| ICD-9-CM | 330.0 |
| DiseasesDB | 32504 |
Leukodystrophy is one of a group of disorders characterized by degeneration of the white matter in the brain.[1] The word leukodystrophy comes from the Greek roots leuko, "white", dys, "lack of", and troph, "growth". The leukodystrophies are caused by imperfect growth or development of the myelin sheath, the fatty covering that acts as an insulator around nerve fibers.
When damage occurs to white matter, immune responses can lead to inflammation in the CNS, along with loss of myelin. The degeneration of white matter can be seen in a MRI and used to diagnose leukodystrophy. Leukodystrophy is characterized by specific symptoms including decreased motor function, muscle rigidity, and eventually degeneration of sight and hearing. While the disease is fatal, the age of onset is a key factor as infants are given a lifespan of 2–8 years (sometimes longer), while adults typically live more than a decade after onset. There is a great lack of treatment, although cord blood and hematopoietic stem cell transplantation (bone marrow transplant) seem to help in certain types while further research is being done.
The combined incidence of the leukodystrophies is estimated at 1:7,600.[2] The majority of types involve the inheritance of a recessive, dominant, or X-linked trait, while others, although involving a defective gene, are the result of spontaneous mutation rather than genetic inheritance.
Types
Specific types of leukodystrophies include the following with their respective ICD-10 codes when available:
- (E71.3) Adrenomyeloneuropathy
- (E75.2) Alexander disease
- (E75.5) Cerebrotendineous xanthomatosis
- Hereditary CNS demyelinating disease
- (E75.2) Krabbe disease
- (E75.2) Metachromatic leukodystrophy
- (E75.2) Pelizaeus-Merzbacher disease
- (E75.2) Canavan disease
- (G93.49) Leukoencephalopathy with vanishing white matter
- (E71.3) Adrenoleukodystrophy
- (G60.1) Refsum disease
- (G70.3) Xenobefantosis
Symptoms and clinical features
Some specific symptoms vary from one type of leukodystrophy to the next but the vast majority of symptoms are shared as the causes for the disease generally have the same effects. Symptoms are dependent on the age of onset, which is predominantly in infancy and early childhood, although the exact time of onset may be difficult to determine. Hyperirritability and hypersensitivity to the environment are common, as well as some tell-tale physical signs including muscle rigidity and a backwards-bent head.[3] Botox therapy is often used to treat patients with spasticity.[4] Juvenile and adult onsets display similar symptoms including a decrease or loss in hearing and vision. While children do experience optic and auditory degeneration, the course of the disease is usually too rapid, causing death relatively quickly, whereas adults may live with these conditions for many years. In children, spastic activity often precedes progressive ataxia and rapid cognitive deterioration which has been described as mental retardation.[5] Epilepsy is commonplace for patients of all ages.[6] More progressed patients show weakness in deglutition, leading to spastic coughing fits due to inhaled saliva. Classic symptomatic progression of juvenile x-linked adrenoleukodystrophy is shown in the 1992 film, Lorenzo's Oil.
Course and timetable are dependent on the age of onset with infants showing a lifespan of 2–8 years, juveniles 2–10 years and adults typically 10+ years. Adults typically see an extended period of stability followed by a decline to a vegetative state and death.[3] While treatments do exist, most are in the experimental phase and can only promise a halt in the progression of symptoms, although some gene therapies have shown some symptomatic improvement.[7] The debilitating course of the disease has led to numerous philosophical and ethical arguments over experimental clinical trials, patients’ rights and physician-assisted suicide.[8]
Causes
While the more specific underlying causes of leukodystrophy are dependent upon the type, there are, however, common pathophysiological patterns that can be seen amongst all types. First and foremost, leukodystrophy is a neurodegenerative disease that is always the result of both impairment and maintenance of myelin sheaths surrounding neuronal axons in the central nervous system as the result of a genetic mutation.[9] Myelin is a fatty white substance that acts as an electrical insulator and coats axons in order to speed up impulses (i.e. action potentials) traveling down the axon. Thus, the natural result of a loss of this substance is decreased efficiency in impulse propagation. As myelin is produced by oligodendrocytes (a type of glial cell) in the central nervous system, an easy place to look for the cause is a mutation or malfunctioning of these cells and in other glial cells.
Genetic influence
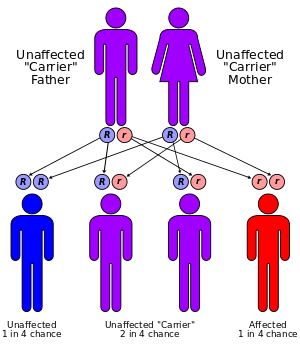
Leukodystrophy is most often an inherited disease that is usually the result of an autosomal recessive inheritance pattern, although dominant inheritance patterns are not unheard of, as in the case of adult-onset leukodystrophy.[10] This means that the affected allele is carried on an autosomal, or non-sex, chromosome and is masked by the dominant, unaffected phenotype. In other words, for an individual to inherit the leukodystrophy phenotype, he or she must carry two of the recessive, mutant alleles. Krabbe Disease and Metachromatic leukodystrophy (MLD) are two of such type. MLD is found on human chromosome 22 at position q13.31.[11] Another type of inherited leukodystrophy is X-linked adrenoleukodystrophy (X-ALD). As its name implies, this type of leukodystrophy is the result of a mutation found on the X-chromosome. It is also carried in a recessive pattern. The X chromosome is a sex chromosome, and since women have two “chances” of acquiring a normal X chromosome (one maternal, one paternal), and males only one (one maternal), this disease is more likely to be seen in men than in women. The mutation resulting in adult-onset leukodystrophy is mapped at 5q23.[10]
Pathophysiology
Although there are nearly forty different types of leukodystrophies, many are lacking in formal and comprehensive research. Most of the research so far has been done on five types: (1) Metachromatic Leukodystrophy (MLD), (2) Krabbe Disease, (3) X-Linked adrenoleukodystrophy (ALD), (4) Canavan Disease, and (5) Alexander Disease. Each type of leukodystrophy has a unique pathophysiology, but all five of these in some way affect a subset of glial cells, therefore disrupting myelin production and maintenance, and usually involve a mutation involving genes that code for enzymes necessary for the catabolism of very long chain fatty acids (VLCFAs) that are toxic to the myelin-producing cells of the central nervous system.[12]
Metachromatic leukodystrophy
Metachromatic leukodystrophy is the result of genetic defects in the enzymes associated with the cellular compartment the lysosome. MLD is inherited in an autosomal recessive way and is the result of mutations in three different ARSA alleles that encode the enzyme arylsulfatase A (ASA), also called sulfatide sulfatase.[13] ASA is responsible for the breakdown of sulfatides, sphingolipids present in neuronal membranes as well as in myelin. When there is a mutation in the gene that encodes ASA, the result is it decreases production, which subsequently leads to diminished degradation of sulfatides, thus causing them to accumulate.[13] This accumulation of sulfatides is poisonous to oligodendrocytes, the myelin-producing cells of the CNS, effectively leading to a disturbance in myelin structure followed by demyelination. The pattern of inheritance of the three different alleles affects what type of MLD a person develops. Two null alleles are responsible for the infantile version, and do not allow for any production of ASA. A heterozygous individual (one null allele, one non-null allele) develops the juvenile form and sees some production of ASA, while an individual with two non-null alleles (but still mutated) develops the adult form.[14]
Krabbe disease
Like MLD, Krabbe disease is another type of leukodystrophy with autosomal recessive inheritance that is the result of a lysosomal storage disorder. It is due to a deletion in exon 16 of the GALC gene that causes a frameshift mutation leading to a premature stop codon. The GALC gene, found on chromosome 14 at position 31 (14q31), codes for the enzyme beta-galactocerebrosidase (GALC).[15] GALC is a lysosomal enzyme responsible for the catabolism of galactolipids, especially psychosine, that are heavily distributed throughout the brain. A deficiency in GALC thus causes a buildup of these fatty acids known as “globoid” macrophages that destroy oligodendrocytes, thereby inhibiting myelin formation.[16] Because of the presence of these globoid cells, clustered near white matter, Krabbe Disease often goes by the name Globoid Cell Leukodystrophy. Furthermore, new research has shown that Krabbe Disease and Globoid Cell Leukodystrophy may be distinct disease entities due to the secretion of inflammatory mediators by natural killer cells in some cases.[17] This research has shown that Natural Killer cells have receptors (TDAG8) for certain glycosphingolipids that build up in an individual with leukodystrophy, again due to insufficient GALC levels, and when bound, target the Natural Killer cells for destruction thereby preventing their cytotoxic effects. These sphingolipids have been identified as galactosyl sphingosine and glycosyl sphingosine and are not present in unaffected individuals.[17]
Canavan disease
Canavan disease is a lesser-studied type of leukodystrophy that, like MLD and Krabbe Disease, is also passed on in an autosomal recessive inheritance pattern. It is due to a mutation in the ASPA gene that encodes aspartoacylase, an enzyme needed to metabolize N-acetyl-L-aspartate (NAA). The mutation causes a deficiency of aspartoacyclase. NAA is involved in the formation of lipids, and if it is not broken down by aspartoacylase, excess levels of it build up causing demyelination.[18]
X-linked adrenoleukodystrophy
In the X-linked adrenoleukodystrophy (X-ALD), a mutation occurs in the peroxisomal ATP-binding cassette (ABC transporter). This leads to cerebral inflammatory demyelination caused by the myelin destabilization that occurs in these patients.[19] The inflammatory demyelination begins in the corpus callosum and it slowly progresses outwards towards both hemispheres. In X-ALD patients, abnormally high levels of very long chain fatty acid (VLCFA) accumulate in various body tissues and fluids. This increased concentration then incorporates into various complex lipids where they are not normally found.[19] This has been found to be directly involved in the cerebral inflammation. The accumulated and embedded VLCFA in the complex lipids could lead to the destabilization of myelin sheath and eventually to demyelination.
Alexander disease
Alexander disease is unique from the leukodystrophies mentioned above in that it is the result of spontaneous mutation, that is it is not inherited. This means that the mutation found in the infected individual is not found in either of his or her parents. It is due to the accumulation of Glial fibrillary acidic protein (GFAP) as the result of a mutation in the GFAP gene; which, rather than being found in association with lysosomes or peroxisomes, is an intermediate filament linked to the nuclear envelope.[20] Intermediate filaments are proteins responsible for the makeup of the cellular cytoskeleton, and thus this type of mutation is involved in malfunctioning structural development of the cells. In fact, cytoskeletal and transporter molecule defects have been observed in the astrocytes (type of glial cell) of affected individuals. These astrocytes have an unhealthily large amount of GFAP that affects astrocyte formation and function.[21]
Epidemiology
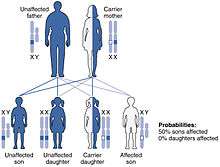
Currently, no research has shown a higher prevalence of most leukodsytrophy types in any one place around the world. There is, however, a higher prevalence of the Canavan disease in the Jewish population for unknown reasons. 1 in 40 individuals of Ashkenazi Jewish descent are carriers of Canavan disease.[22] This estimates to roughly 2.5%. Additionally, due to an autosomal recessive inheritance patterns, there is no significant difference found between affected males and affected females for most types of leukodystrophy including, but not limited to, metachromatic leukodystrophy, Krabbe disease, Canavan disease, and Alexander disease. The one exception to this is any type of leukodystrophy carried on a sex chromosome, such as X-linked adrenoleukodystrophy, which is carried on the X-chromosome. Because of the inheritance pattern of X-linked diseases, males are more often affected by this type of leukodystrophy, although female carriers are often symptomatic, though not as severely so as males.[23] To date, there have been no found cases of a leukodystrophy carried on the Y chromosome.
Diagnosis
The degeneration of white matter, which shows the degeneration of myelin, can be seen in a basic MRI and used to diagnose leukodystrophies of all types. T-1 and T-2 weighted FLAIR images are the most useful. FLAIR stands for fluid-attenuated inversion recovery.[24] Electrophysiological and other kinds of laboratory testing can also be done. In particular, nerve conduction velocity is looked at to distinguish between leukodystrophy and other demyelinating diseases, as well as to distinguish between individual leukodystrophies. For example, individuals with X-ALD have normal conduction velocities, while those with Krabbe disease or metachromatic leukodystrophy have abnormalities in their conduction velocities.[24]
Treatment
With many different types of leukodystrophies and causes, treatment therapies vary for each type. Many studies and clinical trials are in progress to find treatment and therapies for each of the different leukodystrophies. Stem cell transplants and gene therapy appear to be the most promising in treating all leukodystrophies providing it is done as early as possible. For hypomyelinating leukodystrophies, therapeutic research into cell-based therapies appears promising. Oligodendrocyte precursor cells and neural stem cells have been transplanted successfully and have shown to be healthy a year later. Fractional anisotropy and radial diffusivity maps showed possible myelination in the region of the transplant.[25] Induced pluripotent stem cells, oligodendrocyte precursor cells, gene correction, and transplantation to promote the maturation, survival, and myelination of oligodendrocytes seem to be the primary routes for possible treatments.[25]
For three types of leukodystrophies (X-linked adrenoleukodystrophy (X-ALD), metachromatic leukodystrophy (MLD) and Krabbe Disease (globoid cell leukodystrophy - GLD), gene therapy using autologous hematopoietic stem cells to transfer the disease gene with lentiviral vectors have shown to be successful and are currently being used in clinical trials for X-ALD and MLD.[7] The progression of X-ALD has shown to be disrupted with hematopoietic stem cell gene therapy but the exact reason why demyelination stops and the amount of stem cells needed is unclear.[7] While there is an accumulation of very long chain fatty acids in the brain, it does not seem to be the reason behind the disease as gene therapy does not correct it.[7]
Adeno-associated vectors have also been used in intracerebral injections to treat MLD. In some patients with MLD, their IQ increased, nerve conduction improved, their MRIs appeared stable, and had normal enzyme levels.[7] Although the greater majority of patients seem to improve after the transplant, some do not respond well to treatment, which may cause devastating outcomes. For those leukodystrophies that result from a deficiency of lysozyme enzymes, such as Krabbes disease, enzyme replacement therapy seems hopeful, however, this proves difficult as the blood-brain barrier severely limits what can pass through into the central nervous system.[7] Due to this obstacle, most research and clinical trials are turning to allogeneic hematopoietic stem cell transplantation.
Current research
MLD Foundation provides updates on MLD research, including (as of 2015) three clinical trials evaluating gene therapy and enzyme replacement therapy, and various lines of basic research. They are also active in newborn screening.
The Global Leukodystrophy Initiative was formed in 2013 to bring together clinicians, researchers and advocacy groups to focus and improve both clinical care and research.
In addition, many research groups are studying the cellular processes of myelination, which may provide insights into leukodystrophy. Researchers in New York have successfully cured leukodystrophy in mice, using skin cells to repair damaged myelin sheaths. Researchers hypothesize that this treatment may possibly be used in curing human multiple sclerosis.[26]
Public awareness
The World Leukodystrophy Alliance is increasing awareness and working to improve quality of care for the leukodystrophies.
Jill Kelly and her husband, NFL quarterback Jim Kelly, founded Hunter's Hope after their son Hunter (1997-2005) was diagnosed with infantile Krabbe leukodystrophy.[27]
Matthew and Michael Clark of Hull, UK are current sufferers. Their story was the subject of the Channel 4 documentary The Curious Case of the Clark Brothers.[28]
See also
References
- ↑ Sachdev, Perminder S.; Keshavan, Matcheri S. (2010-03-15). Secondary Schizophrenia. Cambridge University Press. pp. 241–. ISBN 978-0-521-85697-3. Retrieved 15 August 2011.
- ↑ Bonkowsky, Joshua (Aug 24, 2010). "The burden of inherited leukodystrophies in children". Neurology. 75 (8): 718–725. doi:10.1212/WNL.0b013e3181eee46b. PMC 2931652
 . PMID 20660364.
. PMID 20660364. - 1 2 Graziano, AC; Cardile, V (26 September 2014). "History, genetic, and recent advances on Krabbe disease". "Gene". 46 (1): 2–13. doi:10.1016/j.gene.2014.09.046. PMID 25260228.
- ↑ "Tardive dystonia and its treatment". Journal of Psychiatry and Neuroscience. 28 (3): 240. PMC 161748.
- ↑ Liu, Y; Zou, L; Meng, Y; Zhang, Y; Shi, X; Ju, J; Yang, G; Hu, L; Chen, X (June 2014). "[A family with two children diagnosed with aspartylglucosaminuria-case report and literature review].". Zhonghua Er Za Zhi. 52: 455–9. PMID 25190167.
- ↑ Turon-Vinas, E; Pineda, M; Cusi, V; Lopez-Laso, E; Del Pozo, RL; Gutierez-Solana, LG; Moreno, DC; Sierra-Corcoles, C; Olabarrieta-Hoyos, N; Madruga-Garrido, M; Aguirre-Rodriguez, J; Gonzalez-Alvarez, V; O'Callaghan, M; Muchart, J; Armstrong-Moron, J (13 July 2014). "Vanishing white matter disease in a Spanish population.". "J Cent Nerv Syst Dis." (6): 59–68.
- 1 2 3 4 5 6 Biffi, Alessandra; Aubourgh, Patrick; Cartier, Nathalie (March 31, 2011). ""Human Molecular Genetics." Gene Therapy for Leukodystrophies". Oxford Journals.
- ↑ Duchange, N; Darguy, S; d'Audiffret, D; Callies, I; Lapointe, AS; Loeve, B; Boespflug-Tanguy, O; Moutel, G (18 September 2014). "Ethical management in the constitution of a European database for leukodystrophies rare diseases.". "Eur J Paediatr Neurol.". 10 (5): 597–603. doi:10.1016/j.ejpn.2014.04.002. PMID 24786336.
- ↑ Yang, Edward; Prabhu, Sanjay P. (March 5, 2014). "Imaging manifestations of the leukodystrophies, inherited disorders of white matter.". Radiologic Clinics of North America. 52 (2): 279–319. doi:10.1016/j.rcl.2013.11.008. PMID 24582341.
- 1 2 Lin, Shu-Ting; Ptacek, Louis J.; Fu, Ying-Hui (January 26, 2011). "Adult-Onset Autosomal Dominant Leukodystrophy: Linking Nuclear Envelope to Myelin". The Journal of Neuroscience. 31 (4): 1163–1166. doi:10.1523/jneurosci.5994-10.2011.
- ↑ Coulter-Mackie, MB; Rip, J; Ludman, MD; Beis, J; Cole, DEC (October 1995). "Metachromatic leucodystrophy (MLD) in a patient with a constitutional ring chromosome 22". Journal of Medical Genetics. 32 (10): 787–91. doi:10.1136/jmg.32.10.787. PMC 1051701. PMID 8558556.
- ↑ Sassa, Takayuki; Kihara, Akio (March 22, 2014). "Metabolism of Very Long-Chain Fatty Acids: Genes and Pathophysiology". Biomolecules and Therapeutics. 22 (2): 83–92. doi:10.4062/biomolther.2014.017. PMC 3975470. PMID 24753812.
- 1 2 Barboura, Ilhem; Ferchichi, Salima; Dandana, Azza; Jaidane, Zaineb; Ben Khelifa, Souhaira; Chahed, Hinda; Ben Mansour, Rachida; Chebel, Saber; Maire, Irene; Miled, Abdelhedi (2010). "Metachromatic leucodystrophy. Clinical, biological, and therapeutic aspects". Annales de Biologie Clinique. 68 (4): Abstract. doi:10.1684/abc.2010.0448. PMID 20650733.
- ↑ Gieselman, V; Krageloh-Mann, I (2010). "Metachromatic Leukodystrophy - An Update". Neuropediatrics. 41 (1): Abstract. doi:10.1055/s-0030-1253412. PMID 20571983.
- ↑ Szymanska, Krystyna; Lugowska, Agnieszka; Laure-Kamionowska, Milena; Gieruszczak-Bialek, Dorota; Musielak, Malgorzata; Eichler, Sabrina; Giese, Anne-Katrin; Rolfs, Arndt (2012). "Diagnostic difficulties in Krabbe disesase: a report of two cases and review of literature". Folia Neuropathol. 50 (4): 346–356. PMID 23319190.
- ↑ Kohlschutter, Alfried (April 25, 2013). "Lysosomal leukodystrophies - Krabbe disease and metachromatic leukodystrophy". Handbook of Clinical Neurology. 113 (Pediatric Neurology Part III): 1611–1618. doi:10.1016/B978-0-444-59565-2.00029-0. Retrieved March 30, 2015.
- 1 2 Maghazachi, Azzam A. (February 5, 2013). "On the Role of Natural Killer Cells in Neurodegenerative Diseases". Toxins (Basel). 5 (2): 363–375. doi:10.3390/toxins5020363. PMC 3640540. PMID 23430541.
- 1 2 Berger, J; Forss-Petter, S; Eichler, F.S. (March 2014). "Pathophysiology of X-Linked Adrenoleukodystrophy". Biochimie. 98: 135–142. doi:10.1016/j.biochi.2013.11.023. PMC 3988840. PMID 24316281.
- ↑ Singh, Navneet; Bixby, Catherine; Etienne, Denzil; Tubbs, R. Shane; Loukas, Marios (December 2012). "Alexander's disease: reassessment of a neonatal form". Child's Nervous System. 28 (12): 2029–2031. doi:10.1007/s00381-012-1868-8. PMID 22890470. Retrieved March 30, 2015.
- ↑ Hol, Elly M.; Pekny, Milos (February 2015). "Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system". Current Opinion in Cell Biology. 32 (Cell Architecture): 121–130. doi:10.1016/j.ceb.2015.02.004. Retrieved March 30, 2015.
- ↑ Lesca, G; Vanier, MT; Creisson, E; Bendelac, N; Hainque, B; Ollagnon-Roman, E; Aubourg, P (August 2005). "X-linked adrenoleukodystrophy in a female proband: clinical presentation, biological diagnosis and family consequences". Archives de Pédiatrie. 12 (8): Abstract. doi:10.1016/j.arcped.2005.03.050. PMID 15878823.
- 1 2 Kohlschutter, Alfried; Eichler, Florian (October 2011). "Childhood leukodystrophies: a clinical perspective". Expert Review of Neurotherapeutics. 11 (10): 1485–1496. doi:10.1586/ern.11.135. Retrieved March 30, 2015.
- 1 2 Pouwels, P. J. W.; Vanderver, A.; Bernard, G.; Wolf, N.; Dreha-Kulczewski, S. W.; Deoni, S. C. L.; Bertini, E.; Kohlschutter, A.; Richardson, W.; ffrench-Constant, C.; Kohler, W.; Barkovich, A. (2014). "Hypomyelinating Leukodystrophies: Translational Research Progress and Prospects". Ann. Neurol. 76: 5–19. doi:10.1002/ana.24194.
- ↑ "Human Skin Cells Used to Create Stem Cells, Treat Brain Disease in Mice". DailyTech. 8 February 2013. Retrieved 9 February 2013.
- ↑ Staff report (October 25, 2012). Game show winners donate portion to Hunter’s Hope. Buffalo News
- ↑ "The Curious Case of the Clark Brothers". Retrieved 2012-11-26.
- This article incorporates public domain text from the National Institute of Neurological Disorders and Stroke.
External links
- Leukodystrophy at DMOZ
- United Leukodystrophy Foundation
- Leukodystrophy Alliance
- MLD Foundation - metachromatic leukodsytrophy
- ALD LIfe - Adrenoleukodsytrophy
- UK Documentary Concerning the Clarke Brothers