X-linked ichthyosis
| X-linked ichthyosis | |
|---|---|
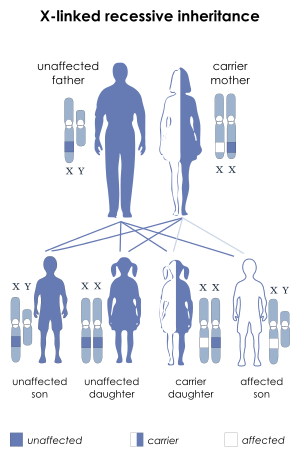 | |
| X-linked recessive inheritance: Affected boys may inherit a deletion or mutation of the STS gene from their mothers | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | Q80.1 |
| ICD-9-CM | 757.1 |
| OMIM | 308100 |
| DiseasesDB | 29136 |
| eMedicine | derm/191 |
| MeSH | D016114 |
| Orphanet | 461 |
X-linked ichthyosis (XLI) (also known as "Steroid sulfatase deficiency," and "X-linked recessive ichthyosis"[1]) (from the Ancient Greek 'ichthys' meaning 'fish') is a skin condition caused by the hereditary deficiency of the steroid sulfatase (STS) enzyme that affects 1 in 2000 to 1 in 6000 males.[2] XLI manifests with dry, scaly skin[3] and is due to deletions[4][5] or mutations[6] in the STS gene. XLI can also occur in the context of larger deletions causing contiguous gene syndromes.[4] Treatment is largely aimed at alleviating the skin symptoms.[7]
Symptoms
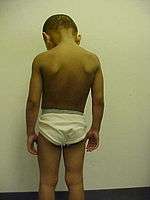
The major symptoms of XLI include scaling of the skin, particularly on the neck, trunk, and lower extremities. The extensor surfaces are typically the most severely affected areas. The >4 mm diameter scales adhere to the underlying skin and can be dark brown or gray in color. Symptoms may subside during the summer.[2]
Genetics
The STS gene is located on the X chromosome at band Xp22.3. Thus, the syndrome is an X-linked condition, and it affects males and females differently. The 23rd pair of chromosomes is typically termed the "sex chromosomes". Females have two X chromosomes and males have one X and one Y chromosome. Therefore, in normal individuals, males carry a single copy of the STS gene and females carry two copies. This gene partially escapes X-inactivation and females normally express higher amounts of the STS enzyme than males.[8]
XLI can occur through new deletions or mutations of the STS gene but is more commonly inherited from a carrier mother.[9] A hemizygous deletion or mutation of the STS gene in a male results in complete absence of enzyme activity, while a female carrier of a mutation or deletion is heterozygous and still has a normal copy of the STS gene. Female carriers of an STS deletion or mutation still express the STS enzyme, although with decreased enzyme activity.[10]
For this reason, XLI most commonly affects males, although individuals with numeric abnormalities of the sex chromosomes (45,X and 47,XXY) who also carry STS deletions or mutations would be exceptions to this rule.
In addition, a female could be affected if she were the offspring of an affected male and a carrier female and inherited a deletion or mutation of the STS gene on both X chromosomes.
Genetic counseling issues
Since the majority of cases appear to occur through transmission of an STS deletion from a carrier mother,[9] enzyme testing or DNA testing should be performed in the mother of any newly diagnosed simplex case (i.e. the first case in a family). In the case of an extended family with many affected individuals, carrier status can often be assigned based on pedigree analysis.
- Males with XLI will transmit the X chromosome harboring the STS deletion or mutation to each of his female offspring, who will therefore be an obligate carrier. However, all male offspring will be unaffected, since they receive their father's Y chromosome.
- Female carriers of an STS deletion or mutation have a 50% chance with each pregnancy of transmitting it to an offspring. Thus, each male offspring has a 50% chance of being affected by XLI, while each female offspring has a 50% chance of being a carrier for this condition. Any individual that inherits the mother's normal copy of the STS gene will be unaffected and will have an extremely low chance of having a child affected with this condition.
Due to random segregation of the chromosomes during gametogenesis, each pregnancy will be subject to the same probabilities, regardless of the number of previously affected or unaffected offspring. It should be noted that the above recurrence risks are based on the assumption that an affected male or carrier female will have children with an unaffected or non-carrier individual. The risks of having affected offspring would clearly increase in the case of a union between a male with XLI and a carrier female.
Associated medical conditions
Aside from the skin scaling, XLI is not typically associated with other major medical problems.[11] Corneal opacities may be present but do not affect vision. Cryptorchidism is reported in some individuals.[2] Some individuals can also be seen to have an intellectual disability, this is thought to be due to deletions encompassing neighboring genes in addition to STS.[12] Larger deletions that include the SHOX gene can result in short stature, while deletions that include the KAL1 gene lead to hypogonadotrophic hypogonadism as seen in Kallmann syndrome.[13]
Female carriers generally do not experience any of these problems but rarely can have difficulty during childbirth, as the STS expressed in the placenta plays a role in normal labor.[14] For this reason carriers should ensure their obstetrician is aware of the condition.
Physiology/biochemistry
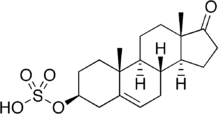
The STS enzyme (EC 3.1.6.2), also referred to as Arylsulfatase C, is expressed throughout the body, with highest expression in the skin, liver, lymph nodes, and placenta, and lower expression in breast tissue and brain[15] STS catalyzes the hydrolysis of sulfated steroids, such as estrone sulfate and dehydroepiandrosterone sulfate (DHEAS), to non-sulfated steroids estradiol and androstenediol, respectively.[16] Prenatally, the enzyme is involved in placental estrogen production.[17] The enzyme is also involved in adrenal steroid production as well as conversion of sulfated steroids in other tissues.
There seems to be a particularly important role for the enzyme in skin. Deficiency of the enzyme leads to the characteristic dry and scaly skin seen in ichthyosis. Recent research indicates that the skin abnormalities seen in XLI may be due to accumulation of cholesterol sulfate in the outer epidermis, leading to abnormal barrier function and corneocyte retention.[18]
Diagnosis
XLI can be suspected based on clinical findings, although symptoms can take varying amounts of time to become evident, from a few hours after birth, up to a year in milder cases. The diagnosis is usually made by a dermatologist, who also typically formulates the treatment plan (see below). STS enzyme deficiency is confirmed using a clinically available biochemical assay. Carrier detection can be performed in mothers of affected sons using this test (see Genetics, below).[10] Molecular testing for DNA deletions or mutations is also offered, and can be particularly useful in the evaluation of individuals with associated medical conditions (see below). Prenatal diagnosis is possible using either biochemical or molecular tests. However, the use of prenatal diagnosis for genetic conditions that are considered to be generally benign raises serious ethical considerations and requires detailed genetic counseling.
Treatment
Because XLI is caused by a gene mutation or deletion, there is no "cure." One of the aims of treatment is to reduce scaling by removing the excess, flaky scales, and keep the skin hydrated. This can be achieved using a variety of topical creams.[2][7]
- Keratolytic agents such as Ammonium lactate (Lac-Hydrin) are used to facilitate the release of retained corneocytes.
- Topical isotretinoin
- The topical receptor-selective retinoid tazarotene [19]
Research is ongoing with regard to the use of gene therapy to treat XLI.[20]
History
In the 1960s, recessive x-linked ichthyosis was distinguished clinically from other ichthyoses.[21]:486[22]:561
See also
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- 1 2 3 4 Carlo Gelmetti; Caputo, Ruggero (2002). Pediatric Dermatology and Dermatopathology: A Concise Atlas. T&F STM. p. 160. ISBN 1-84184-120-X.
- ↑ Online Mendelian Inheritance in Man (OMIM) ICHTHYOSIS, X-LINKED -308100
- 1 2 Ballabio A, Parenti G, Carrozzo R, et al. (1987). "Isolation and characterization of a steroid sulfatase cDNA clone: genomic deletions in patients with X-chromosome-linked ichthyosis". Proc. Natl. Acad. Sci. U.S.A. 84 (13): 4519–23. doi:10.1073/pnas.84.13.4519. PMC 305121
 . PMID 3474618.
. PMID 3474618.
- ↑ Bonifas JM, Morley BJ, Oakey RE, Kan YW, Epstein EH (December 1987). "Cloning of a cDNA for steroid sulfatase: frequent occurrence of gene deletions in patients with recessive X chromosome-linked ichthyosis". Proc. Natl. Acad. Sci. U.S.A. 84 (24): 9248–51. doi:10.1073/pnas.84.24.9248. PMC 299730. PMID 3480541.
- ↑ Basler E, Grompe M, Parenti G, Yates J, Ballabio A (March 1992). "Identification of point mutations in the steroid sulfatase gene of three patients with X-linked ichthyosis". Am. J. Hum. Genet. 50 (3): 483–91. PMC 1684279. PMID 1539590.
- 1 2 Ichthyosis, X-Linked at eMedicine: Treatment Section
- ↑ Lykkesfeldt G, Lykkesfeldt AE, Skakkebaek NE (1984). "Steroid sulphatase in man: a non inactivated X-locus with partial gene dosage compensation". Hum. Genet. 65 (4): 355–7. doi:10.1007/BF00291559. PMID 6582028.
- 1 2 Valdes-Flores M, Kofman-Alfaro SH, Jimenez-Vaca AL, Cuevas-Covarrubias SA (August 2001). "Carrier identification by FISH analysis in isolated cases of X-linked ichthyosis". Am. J. Med. Genet. 102 (2): 146–8. doi:10.1002/ajmg.1450. PMID 11477606.
- 1 2 Cuevas-Covarrubias SA, Kofman-Alfaro S, Orozco Orozco E, Diaz-Zagoya JC (1995). "The biochemical identification of carrier state in mothers of sporadic cases of X-linked recessive ichthyosis". Genet. Couns. 6 (2): 103–7. PMID 7546451.
- ↑ DiGiovanna JJ, Robinson-Bostom L (2003). "Ichthyosis: etiology, diagnosis, and management". Am J Clin Dermatol. 4 (2): 81–95. doi:10.2165/00128071-200304020-00002. PMID 12553849.
- ↑ Van Esch H, Hollanders K, Badisco L, et al. (2005). "Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis". Hum. Mol. Genet. 14 (13): 1795–803. doi:10.1093/hmg/ddi186. PMID 15888481.
- ↑ Ballabio A, Sebastio G, Carrozzo R, et al. (1987). "Deletions of the steroid sulphatase gene in "classical" X-linked ichthyosis and in X-linked ichthyosis associated with Kallmann syndrome". Hum. Genet. 77 (4): 338–41. doi:10.1007/BF00291422. PMID 3480263.
- ↑ Bradshaw KD, Carr BR (1986). "Placental sulfatase deficiency: maternal and fetal expression of steroid sulfatase deficiency and X-linked ichthyosis". Obstet Gynecol Surv. 41 (7): 401–13. PMID 3531932.
- ↑ Selcer KW, Difrancesca HM, Chandra AB, Li PK (2007). "Immunohistochemical analysis of steroid sulfatase in human tissues". J. Steroid Biochem. Mol. Biol. 105 (1-5): 115–23. doi:10.1016/j.jsbmb.2006.12.105. PMID 17604157.
- ↑ Reed MJ, Purohit A, Woo LW, Newman SP, Potter BV (April 2005). "Steroid sulfatase: molecular biology, regulation, and inhibition". Endocr. Rev. 26 (2): 171–202. doi:10.1210/er.2004-0003. PMID 15561802.
- ↑ Jöbsis AC, De Groot WP, Tigges AJ, et al. (1980). "X-linked ichthyosis and X-linked placental sulfatase deficiency: a disease entity. Histochemical observations". Am. J. Pathol. 99 (2): 279–89. PMC 1903491. PMID 6929654.
- ↑ Elias PM, Crumrine D, Rassner U, et al. (2004). "Basis for abnormal desquamation and permeability barrier dysfunction in RXLI". J. Invest. Dermatol. 122 (2): 314–9. doi:10.1046/j.1523-1747.2003.22258.x. PMID 15009711.
- ↑ Cotellessa C, Cuevas-Covarrubias SA, Valeri P, Fargnoli MC, Peris K (2005). "Topical tazarotene 0.05% versus glycolic acid 70% treatment in X-linked ichthyosis due to extensive deletion of the STS gene". Acta Derm. Venereol. 85 (4): 346–8. doi:10.1080/00015550510026613. PMID 16191859.
- ↑ Freiberg RA, Choate KA, Deng H, Alperin ES, Shapiro LJ, Khavari PA (1997). "A model of corrective gene transfer in X-linked ichthyosis". Hum. Mol. Genet. 6 (6): 927–33. doi:10.1093/hmg/6.6.927. PMID 9175741.
- ↑ Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
External links
The Ichthyosis Support Group - http://www.ichthyosis.org.uk.