Jervell and Lange-Nielsen syndrome
| Jervell and Lange-Nielsen syndrome | |
|---|---|
| Classification and external resources | |
| ICD-9-CM | 426.82 |
| OMIM | 220400 |
| DiseasesDB | 7249 |
| MeSH | D029593 |
| GeneReviews | |
Jervell and Lange-Nielsen syndrome (JLNS) is a type of long QT syndrome, associated with severe, bilateral sensorineural hearing loss. Long QT syndrome causes the cardiac muscle to take longer than usual to recharge between beats. If untreated, the irregular heartbeats, called arrhythmias, can lead to fainting, seizures, or sudden death. It was first described by Anton Jervell and Fred Lange-Nielsen in 1957.[1]
Genetic prevalence
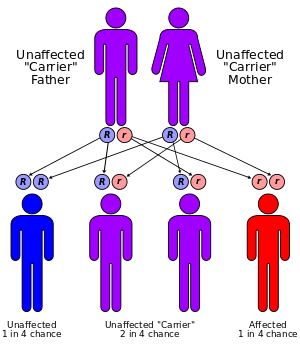
This condition is an autosomal recessive disorder that affects an estimated 1.6 to 6 in 1 million children, and is responsible for less than 10 percent of all cases of long QT syndrome. It has a markedly higher incidence in Norway and Sweden, up to 1:200,000.[2] Mutations in the KCNE1 and KCNQ1 genes cause Jervell and Lange-Nielsen syndrome. The proteins produced by these two genes work together to form a potassium channel that transports positively charged potassium ions out of cells. The movement of potassium ions through these channels is critical for maintaining the normal functions of the inner ear and cardiac muscle.[2]
About 90 percent of cases of Jervell and Lange-Nielsen syndrome are caused by mutations in the KCNQ1 gene. KCNE1 mutations are responsible for the remaining 10 percent of cases. Mutations in these genes alter the usual structure and function of potassium channels or prevent the assembly of normal channels. These changes disrupt the flow of potassium ions in the inner ear and in cardiac muscle, leading to the hearing loss and irregular heart rhythm characteristic of Jervell and Lange-Nielsen syndrome.[2]
Patients with Jervell and Lange-Nielsen syndrome are born with a severe to profound sensorineural hearing loss which is stable. They have very poor vestibular (inner ear balance) function which means that they are slow achieving locomotor milestones such as sitting and walking.
Treatment
JLNS patients with KCNQ1 mutations are particularly prone to pathological lengthening of the QT interval, which predisposes them to episodes of Torsades de pointes and sudden cardiac death. In this context if the patient has had syncopal episodes or history of cardiac arrest, an implantable cardiac defibrillator should be used in addition to a beta blocker such as propranolol.[2]
References
- ↑ Jervell, A.; Lange-Nielsen, F. (1957). "Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death". American Heart Journal. 54 (1): 59–68. doi:10.1016/0002-8703(57)90079-0. PMID 13435203.
- 1 2 3 4 Tranebjaerg, L.; Samson, R.A.; Green, G.E. "Jervell and Lange-Nielsen Syndrome". GeneReviews. Retrieved 29 November 2013.
External links
This article incorporates public domain text from The U.S. National Library of Medicine