Tyrosinemia
| Tyrosinemia | |
|---|---|
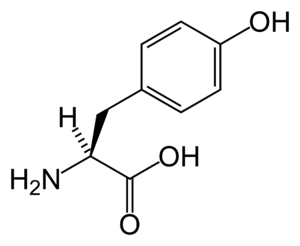 | |
| Tyrosine | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E70.2 |
| ICD-9-CM | 270.2 |
| OMIM | 276700 276600 276710 |
| DiseasesDB | 29836 |
| eMedicine | ped/2339 |
| Patient UK | Tyrosinemia |
| MeSH | D020176 |
Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms include liver and kidney disturbances and intellectual disability. Untreated, tyrosinemia can be fatal.
Most inborn forms of tyrosinemia produce hypertyrosinemia (high levels of tyrosine).[1]
Types
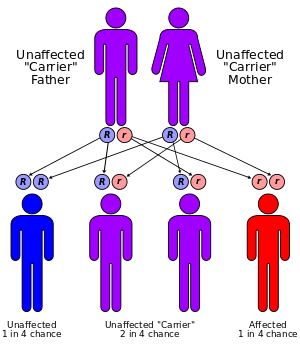
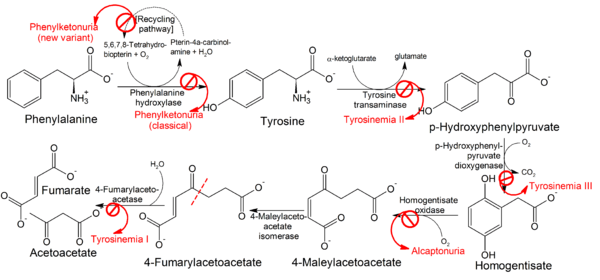
There are three types of tyrosinemia, each with distinctive symptoms and caused by the deficiency of a different enzyme.
Treatment
Treatment varies depending on the specific type. A low protein diet may be required in the management of tyrosinemia. Recent experience with nitisinone has shown it to be effective. It is a 4-hydroxyphenylpyruvate dioxygenase inhibitor indicated for the treatment of hereditary tyrosinemia type 1 (HT-1) in combination with dietary restriction of tyrosine and phenylalanine.[2] The most effective treatment in patients with tyrosinemia type I seems to be full or partial liver transplant.
See also
References
- ↑ Charles Scriver, Beaudet, A.L., Valle, D., Sly, W.S., Vogelstein, B., Childs, B., Kinzler, K.W. (Accessed 2007). The Online Metabolic and Molecular Bases of Inherited Disease. Chapter 79. New York: McGraw-Hill.
- ↑ Swedish Orphan Biovitrum AB, Orfadin [package insert] (PDF), retrieved 2016-07-12
External links
- GeneReview/NCBI/NIH/UW entry on Tyrosinemia Type 1
- University of Washington
- Tyrosinemia on Genetic Home Reference